Introduction to clustifyr
Rui Fu
RNA Bioscience Initative, University of Colorado School of MedicineAustin Gillen
RNA Bioscience Initative, University of Colorado School of MedicineRyan Sheridan
RNA Bioscience Initative, University of Colorado School of MedicineChengzhe Tian
Department of Biochemistry, University of Colorado BoulderMichelle Daya
Biomedical Informatics & Personalized Medicine, University of Colorado Anschutz Medical CampusYue Hao
Bioinformatics Research Center, North Carolina State UniversityJay Hesselberth
RNA Bioscience Initative, University of Colorado School of MedicineKent Riemondy
RNA Bioscience Initative, University of Colorado School of Medicine2022-07-06
Source:vignettes/clustifyR.Rmd
clustifyR.RmdIntroduction: Why use clustifyr?
Single cell transcriptomes are difficult to annotate without extensive knowledge of the underlying biology of the system in question. Even with this knowledge, accurate identification can be challenging due to the lack of detectable expression of common marker genes defined by bulk RNA-seq, flow cytometry, other single cell RNA-seq platforms, etc.
clustifyr solves this problem by providing functions to automatically annotate single cells or clusters using bulk RNA-seq data or marker gene lists (ranked or unranked). Additional functions allow for exploratory analysis of calculated similarities between single cell RNA-seq datasets and reference data.
Installation
To install clustifyr BiocManager must be installed.
install.packages("BiocManager")
BiocManager::install("clustifyr")A simple example: 10x Genomics PBMCs
In this example, we take a 10x Genomics 3’ scRNA-seq dataset from peripheral blood mononuclear cells (PBMCs) and annotate the cell clusters (identified using Seurat) using scRNA-seq cell clusters assigned from a CITE-seq experiment.
library(clustifyr)
library(ggplot2)
library(cowplot)
# Matrix of normalized single-cell RNA-seq counts
pbmc_matrix <- clustifyr::pbmc_matrix_small
# meta.data table containing cluster assignments for each cell
# The table that we are using also contains the known cell identities in the "classified" column
pbmc_meta <- clustifyr::pbmc_metaCalculate correlation coefficients
To identify cell types, the clustifyr() function requires several inputs:
-
input: an SingleCellExperiment or Seurat object or a matrix of normalized single-cell RNA-seq counts -
metadata: a meta.data table containing the cluster assignments for each cell (not required if a Seurat object is given) -
ref_mat: a reference matrix containing RNA-seq expression data for each cell type of interest -
query_genes: a list of genes to use for comparison (optional but recommended)
When using a matrix of scRNA-seq counts clustifyr() will return a matrix of correlation coefficients for each cell type and cluster, with the rownames corresponding to the cluster number.
# Calculate correlation coefficients for each cluster (spearman by default)
vargenes <- pbmc_vargenes[1:500]
res <- clustify(
input = pbmc_matrix, # matrix of normalized scRNA-seq counts (or SCE/Seurat object)
metadata = pbmc_meta, # meta.data table containing cell clusters
cluster_col = "seurat_clusters", # name of column in meta.data containing cell clusters
ref_mat = cbmc_ref, # matrix of RNA-seq expression data for each cell type
query_genes = vargenes # list of highly varible genes identified with Seurat
)
# Peek at correlation matrix
res[1:5, 1:5]
#> B CD14+ Mono CD16+ Mono CD34+ CD4 T
#> 0 0.6563466 0.6454029 0.6485863 0.7089861 0.8804508
#> 1 0.6394363 0.6388404 0.6569401 0.7027430 0.8488750
#> 2 0.5524081 0.9372089 0.8930158 0.5879264 0.5347312
#> 3 0.8945380 0.5801453 0.6146857 0.6955897 0.6566739
#> 4 0.5711643 0.5623870 0.5826233 0.6280913 0.7467347
# Call cell types
res2 <- cor_to_call(
cor_mat = res, # matrix correlation coefficients
cluster_col = "seurat_clusters" # name of column in meta.data containing cell clusters
)
res2[1:5, ]
#> # A tibble: 5 × 3
#> # Groups: seurat_clusters [5]
#> seurat_clusters type r
#> <chr> <chr> <dbl>
#> 1 3 B 0.895
#> 2 2 CD14+ Mono 0.937
#> 3 5 CD16+ Mono 0.929
#> 4 0 CD4 T 0.880
#> 5 1 CD4 T 0.849
# Insert into original metadata as "type" column
pbmc_meta2 <- call_to_metadata(
res = res2, # data.frame of called cell type for each cluster
metadata = pbmc_meta, # original meta.data table containing cell clusters
cluster_col = "seurat_clusters" # name of column in meta.data containing cell clusters
)To visualize the clustifyr() results we can use the plot_cor_heatmap() function to plot the correlation coefficients for each cluster and each cell type.
# Create heatmap of correlation coefficients using clustifyr() output
plot_cor_heatmap(cor_mat = res)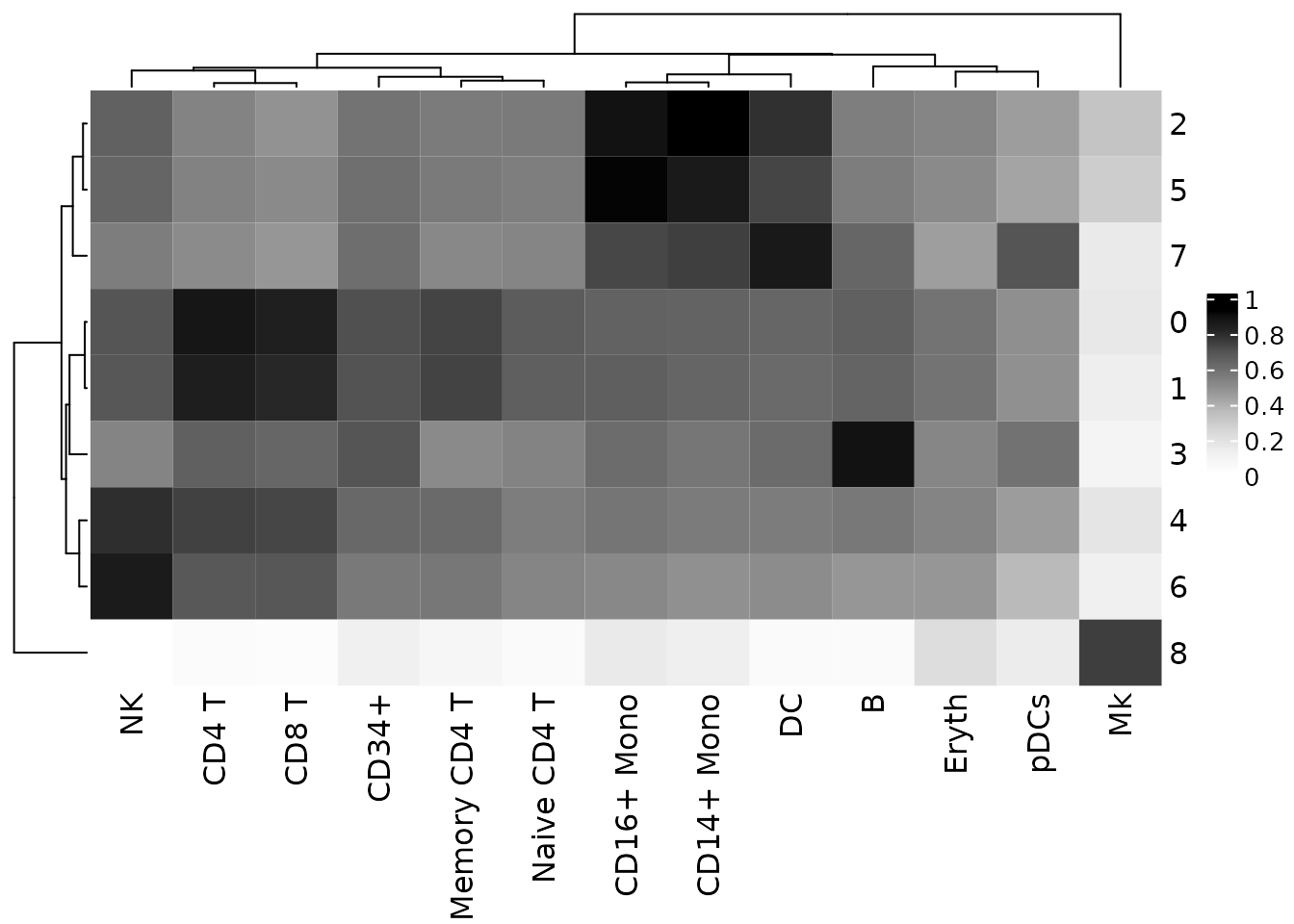
Plot cluster identities and correlation coefficients
clustifyr also provides functions to overlay correlation coefficients on pre-calculated tSNE embeddings (or those from any other dimensionality reduction method).
# Overlay correlation coefficients on UMAPs for the first two cell types
corr_umaps <- plot_cor(
cor_mat = res, # matrix of correlation coefficients from clustifyr()
metadata = pbmc_meta, # meta.data table containing UMAP or tSNE data
data_to_plot = colnames(res)[1:2], # name of cell type(s) to plot correlation coefficients
cluster_col = "seurat_clusters" # name of column in meta.data containing cell clusters
)
plot_grid(
plotlist = corr_umaps,
rel_widths = c(0.47, 0.53)
)
The plot_best_call() function can be used to label each cluster with the cell type that gives the highest corelation coefficient. Using the plot_dims() function, we can also plot the known identities of each cluster, which were stored in the “classified” column of the meta.data table. The plots below show that the highest correlations between the reference RNA-seq data and the 10x Genomics scRNA-seq dataset are restricted to the correct cell clusters.
# Label clusters with clustifyr cell identities
clustifyr_types <- plot_best_call(
cor_mat = res, # matrix of correlation coefficients from clustifyr()
metadata = pbmc_meta, # meta.data table containing UMAP or tSNE data
do_label = TRUE, # should the feature label be shown on each cluster?
do_legend = FALSE, # should the legend be shown?
cluster_col = "seurat_clusters"
) +
ggtitle("clustifyr cell types")
# Compare clustifyr results with known cell identities
known_types <- plot_dims(
data = pbmc_meta, # meta.data table containing UMAP or tSNE data
feature = "classified", # name of column in meta.data to color clusters by
do_label = TRUE, # should the feature label be shown on each cluster?
do_legend = FALSE # should the legend be shown?
) +
ggtitle("Known cell types")
plot_grid(known_types, clustifyr_types)
Classify cells using known marker genes
The clustify_lists() function allows cell types to be assigned based on known marker genes. This function requires a table containing markers for each cell type of interest. Cell types can be assigned using several statistical tests including, hypergeometric, Jaccard, Spearman, and GSEA.
# Take a peek at marker gene table
cbmc_m
#> CD4 T CD8 T Memory CD4 T CD14+ Mono Naive CD4 T NK B CD16+ Mono
#> 1 ITM2A CD8B ADCY2 S100A8 CDHR3 GNLY IGHM CDKN1C
#> 2 TXNIP CD8A PTGDR2 S100A9 DICER1-AS1 NKG7 CD79A HES4
#> 3 AES S100B CD200R1 LYZ RAD9A CST7 MS4A1 CKB
#> CD34+ Eryth Mk DC pDCs
#> 1 SPINK2 HBM PF4 ENHO LILRA4
#> 2 C1QTNF4 AHSP SDPR CD1E TPM2
#> 3 KIAA0125 CA1 TUBB1 NDRG2 SCT
# Available metrics include: "hyper", "jaccard", "spearman", "gsea"
list_res <- clustify_lists(
input = pbmc_matrix, # matrix of normalized single-cell RNA-seq counts
metadata = pbmc_meta, # meta.data table containing cell clusters
cluster_col = "seurat_clusters", # name of column in meta.data containing cell clusters
marker = cbmc_m, # list of known marker genes
metric = "pct" # test to use for assigning cell types
)
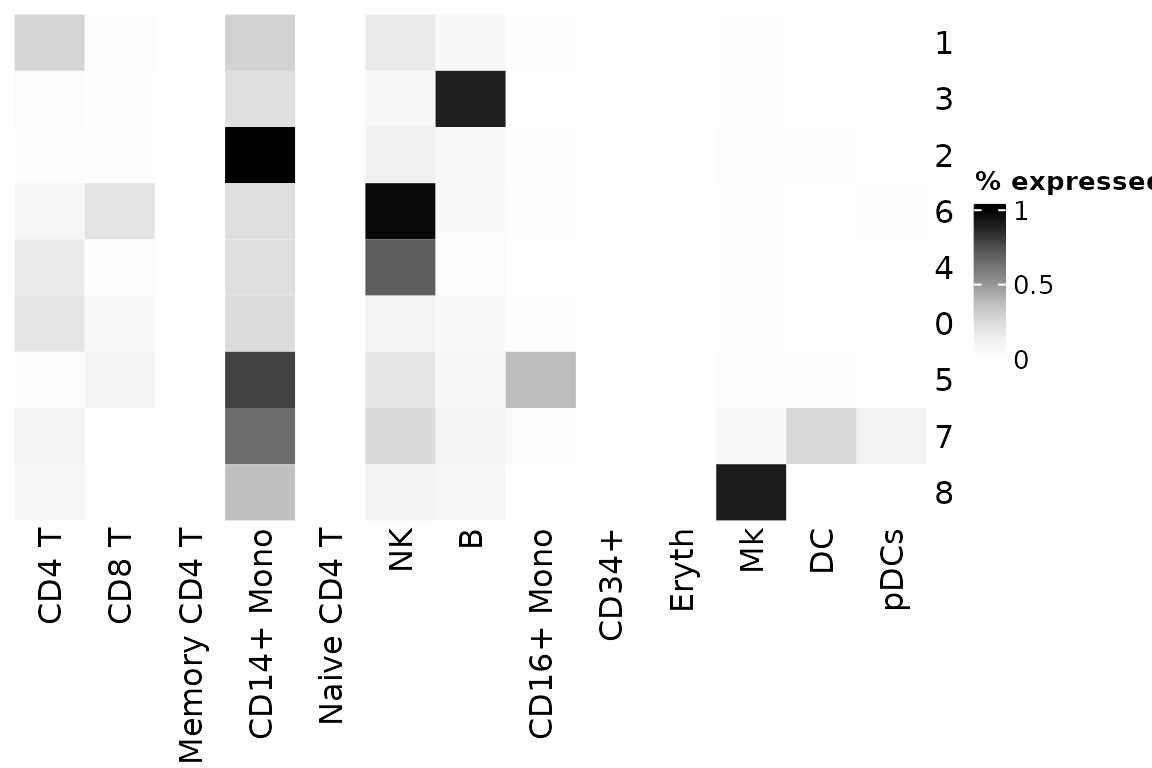
# View as heatmap, or plot_best_call
plot_cor_heatmap(
cor_mat = list_res, # matrix of correlation coefficients from clustify_lists()
cluster_rows = FALSE, # cluster by row?
cluster_columns = FALSE, # cluster by column?
legend_title = "% expressed" # title of heatmap legend
)
# Downstream functions same as clustify()
# Call cell types
list_res2 <- cor_to_call(
cor_mat = list_res, # matrix correlation coefficients
cluster_col = "seurat_clusters" # name of column in meta.data containing cell clusters
)
# Insert into original metadata as "list_type" column
pbmc_meta3 <- call_to_metadata(
res = list_res2, # data.frame of called cell type for each cluster
metadata = pbmc_meta, # original meta.data table containing cell clusters
cluster_col = "seurat_clusters", # name of column in meta.data containing cell clusters
rename_prefix = "list_" # set a prefix for the new column
)Direct handling of SingleCellExperiment objects
clustifyr can also use a SingleCellExperiment object as input and return a new SingleCellExperiment object with the cell types added as a column in the colData.
res <- clustify(
input = sce_small, # an SCE object
ref_mat = cbmc_ref, # matrix of RNA-seq expression data for each cell type
cluster_col = "cell_type1", # name of column in meta.data containing cell clusters
obj_out = TRUE # output SCE object with cell type inserted as "type" column
)
SingleCellExperiment::colData(res)[1:10,c("type", "r")]
#> DataFrame with 10 rows and 2 columns
#> type r
#> <character> <numeric>
#> AZ_A1 CD34+ 0.557678024919381
#> AZ_A10 CD34+ 0.624777701661225
#> AZ_A11 CD4 T 0.695067885340303
#> AZ_A12 CD34+ 0.624777701661225
#> AZ_A2 CD4 T 0.602804908958642
#> AZ_A3 CD34+ 0.557678024919381
#> AZ_A4 CD34+ 0.557678024919381
#> AZ_A5 CD34+ 0.645378073051508
#> AZ_A6 CD4 T 0.695067885340303
#> AZ_A7 CD34+ 0.671644883893203Direct handling of seurat v2 and v3 objects
clustifyr can also use a Seurat object as input and return a new Seurat object with the cell types added as a column in the meta.data.
res <- clustify(
input = s_small3, # a Seurat object
ref_mat = cbmc_ref, # matrix of RNA-seq expression data for each cell type
cluster_col = "RNA_snn_res.1", # name of column in meta.data containing cell clusters
obj_out = TRUE # output Seurat object with cell type inserted as "type" column
)
res@meta.data[1:10, ]
#> nGene nUMI orig.ident res.0.8 res.1 type r
#> ATGCCAGAACGACT 47 70 SeuratProject 0 0 Memory CD4 T 0.7047302
#> CATGGCCTGTGCAT 52 85 SeuratProject 0 0 Memory CD4 T 0.7047302
#> GAACCTGATGAACC 50 87 SeuratProject 0 0 Memory CD4 T 0.7047302
#> TGACTGGATTCTCA 56 127 SeuratProject 0 0 Memory CD4 T 0.7047302
#> AGTCAGACTGCACA 53 173 SeuratProject 0 0 Memory CD4 T 0.7047302
#> TCTGATACACGTGT 48 70 SeuratProject 0 0 Memory CD4 T 0.7047302
#> TGGTATCTAAACAG 36 64 SeuratProject 0 0 Memory CD4 T 0.7047302
#> GCAGCTCTGTTTCT 45 72 SeuratProject 0 0 Memory CD4 T 0.7047302
#> GATATAACACGCAT 36 52 SeuratProject 0 0 Memory CD4 T 0.7047302
#> AATGTTGACAGTCA 41 100 SeuratProject 0 0 Memory CD4 T 0.7047302Building reference matrix from single cell expression matrix
In its simplest form, a reference matrix is built by averaging expression (also includes an option to take the median) of a single cell RNA-seq expression matrix by cluster. Both log transformed or raw count matrices are supported.
new_ref_matrix <- average_clusters(
mat = pbmc_matrix,
metadata = pbmc_meta$classified, # or use metadata = pbmc_meta, cluster_col = "classified"
if_log = TRUE # whether the expression matrix is already log transformed
)
head(new_ref_matrix)
#> B CD14+ Mono CD8 T DC FCGR3A+ Mono
#> PPBP 0.09375021 0.28763857 0.35662599 0.06527347 0.2442300
#> LYZ 1.42699419 5.21550849 1.35146753 4.84714962 3.4034309
#> S100A9 0.62123058 4.91453355 0.58823794 2.53310734 2.6277996
#> IGLL5 2.44576997 0.02434753 0.03284986 0.10986617 0.2581198
#> GNLY 0.37877736 0.53592906 2.53161887 0.46959958 0.2903092
#> FTL 3.66698837 5.86217774 3.37056910 4.21848878 5.9518479
#> Memory CD4 T Naive CD4 T NK Platelet
#> PPBP 0.06494743 0.04883837 0.00000000 6.0941782
#> LYZ 1.39466552 1.40165143 1.32701580 2.5303912
#> S100A9 0.58080250 0.55679700 0.52098541 1.6775692
#> IGLL5 0.04826212 0.03116080 0.05247669 0.2501642
#> GNLY 0.41001072 0.46041901 4.70481754 0.3845813
#> FTL 3.31062958 3.35611600 3.38471536 4.5508242
# For further convenience, a shortcut function for generating reference matrix from `SingleCellExperiment` or `seurat` object is used.
new_ref_matrix_sce <- object_ref(
input = sce_small, # SCE object
cluster_col = "cell_type1" # name of column in colData containing cell identities
)
new_ref_matrix_v3 <- seurat_ref(
seurat_object = s_small3, # SeuratV3 object
cluster_col = "RNA_snn_res.1" # name of column in meta.data containing cell identities
)
tail(new_ref_matrix_v3)
#> 0 1 2
#> C12orf75 3.223784 0.4297757 2.305767
#> RARRES3 4.341699 1.7550597 3.794566
#> PCMT1 4.576310 2.8893691 2.223141
#> LAMP1 3.919129 0.9441022 1.565788
#> SPON2 3.670497 1.3182933 3.200796
#> S100B 3.069210 0.0000000 0.000000
# There's also the option to pull UCSC cell browser data.
get_ext_reference(cb_url = "http://cells.ucsc.edu/?ds=kidney-atlas%2FFetal_Immune",
cluster_col = "celltype")More reference data, including tabula muris, and code used to generate them are available at https://github.com/rnabioco/clustifyrdata
Also see list for individual downloads at https://rnabioco.github.io/clustifyrdata/articles/download_refs.html
Additional tutorials at https://rnabioco.github.io/clustifyrdata/articles/otherformats.html
Session info
sessionInfo()
#> R version 4.2.0 (2022-04-22)
#> Platform: x86_64-pc-linux-gnu (64-bit)
#> Running under: Ubuntu 20.04.4 LTS
#>
#> Matrix products: default
#> BLAS: /usr/lib/x86_64-linux-gnu/blas/libblas.so.3.9.0
#> LAPACK: /usr/lib/x86_64-linux-gnu/lapack/liblapack.so.3.9.0
#>
#> locale:
#> [1] LC_CTYPE=C.UTF-8 LC_NUMERIC=C LC_TIME=C.UTF-8
#> [4] LC_COLLATE=C.UTF-8 LC_MONETARY=C.UTF-8 LC_MESSAGES=C.UTF-8
#> [7] LC_PAPER=C.UTF-8 LC_NAME=C LC_ADDRESS=C
#> [10] LC_TELEPHONE=C LC_MEASUREMENT=C.UTF-8 LC_IDENTIFICATION=C
#>
#> attached base packages:
#> [1] stats graphics grDevices utils datasets methods base
#>
#> other attached packages:
#> [1] cowplot_1.1.1 ggplot2_3.3.6 clustifyr_1.8.0 BiocStyle_2.24.0
#>
#> loaded via a namespace (and not attached):
#> [1] bitops_1.0-7 matrixStats_0.62.0
#> [3] fs_1.5.2 doParallel_1.0.17
#> [5] RColorBrewer_1.1-3 httr_1.4.3
#> [7] rprojroot_2.0.3 GenomeInfoDb_1.32.2
#> [9] tools_4.2.0 bslib_0.3.1
#> [11] utf8_1.2.2 R6_2.5.1
#> [13] BiocGenerics_0.42.0 colorspace_2.0-3
#> [15] GetoptLong_1.0.5 withr_2.5.0
#> [17] tidyselect_1.1.2 gridExtra_2.3
#> [19] compiler_4.2.0 textshaping_0.3.6
#> [21] cli_3.3.0 Biobase_2.56.0
#> [23] desc_1.4.1 DelayedArray_0.22.0
#> [25] labeling_0.4.2 entropy_1.3.1
#> [27] bookdown_0.27 sass_0.4.1
#> [29] scales_1.2.0 pkgdown_2.0.5
#> [31] systemfonts_1.0.4 stringr_1.4.0
#> [33] digest_0.6.29 rmarkdown_2.14
#> [35] XVector_0.36.0 pkgconfig_2.0.3
#> [37] htmltools_0.5.2 MatrixGenerics_1.8.1
#> [39] highr_0.9 fastmap_1.1.0
#> [41] rlang_1.0.3 GlobalOptions_0.1.2
#> [43] farver_2.1.0 shape_1.4.6
#> [45] jquerylib_0.1.4 generics_0.1.2
#> [47] jsonlite_1.8.0 BiocParallel_1.30.3
#> [49] dplyr_1.0.9 RCurl_1.98-1.7
#> [51] magrittr_2.0.3 GenomeInfoDbData_1.2.8
#> [53] Matrix_1.4-1 Rcpp_1.0.8.3
#> [55] munsell_0.5.0 S4Vectors_0.34.0
#> [57] fansi_1.0.3 lifecycle_1.0.1
#> [59] stringi_1.7.6 yaml_2.3.5
#> [61] SummarizedExperiment_1.26.1 zlibbioc_1.42.0
#> [63] grid_4.2.0 ggrepel_0.9.1
#> [65] parallel_4.2.0 crayon_1.5.1
#> [67] lattice_0.20-45 circlize_0.4.15
#> [69] knitr_1.39 ComplexHeatmap_2.12.0
#> [71] pillar_1.7.0 fgsea_1.22.0
#> [73] GenomicRanges_1.48.0 rjson_0.2.21
#> [75] codetools_0.2-18 stats4_4.2.0
#> [77] fastmatch_1.1-3 glue_1.6.2
#> [79] evaluate_0.15 data.table_1.14.2
#> [81] BiocManager_1.30.18 vctrs_0.4.1
#> [83] png_0.1-7 foreach_1.5.2
#> [85] gtable_0.3.0 purrr_0.3.4
#> [87] tidyr_1.2.0 clue_0.3-61
#> [89] cachem_1.0.6 xfun_0.31
#> [91] ragg_1.2.2 SingleCellExperiment_1.18.0
#> [93] tibble_3.1.7 iterators_1.0.14
#> [95] memoise_2.0.1 IRanges_2.30.0
#> [97] cluster_2.1.3 ellipsis_0.3.2