clustifyr classifies cells and clusters in single-cell RNA sequencing experiments using reference bulk RNA-seq data sets, sorted microarray expression data, single-cell gene signatures, or lists of marker genes.
Installation
Install the Bioconductor version with:
if (!requireNamespace("BiocManager", quietly = TRUE))
install.packages("BiocManager")
BiocManager::install("clustifyr")Install the development version with:
BiocManager::install("rnabioco/clustifyr")Example usage
In this example we use the following built-in input data:
- an expression matrix of single cell RNA-seq data (
pbmc_matrix_small) - a metadata data.frame (
pbmc_meta), with cluster information stored ("classified") - a vector of variable genes (
pbmc_vargenes) - a matrix of mean normalized scRNA-seq UMI counts by cell type (
cbmc_ref)
We then calculate correlation coefficients and plot them on a pre-calculated projection (stored in pbmc_meta).
library(clustifyr)
# calculate correlation
res <- clustify(
input = pbmc_matrix_small,
metadata = pbmc_meta$classified,
ref_mat = cbmc_ref,
query_genes = pbmc_vargenes
)
# print assignments
cor_to_call(res)
#> # A tibble: 9 × 3
#> # Groups: cluster [9]
#> cluster type r
#> <chr> <chr> <dbl>
#> 1 B B 0.909
#> 2 CD14+ Mono CD14+ Mono 0.915
#> 3 FCGR3A+ Mono CD16+ Mono 0.929
#> 4 Memory CD4 T CD4 T 0.861
#> 5 Naive CD4 T CD4 T 0.889
#> 6 DC DC 0.849
#> 7 Platelet Mk 0.732
#> 8 CD8 T NK 0.826
#> 9 NK NK 0.894
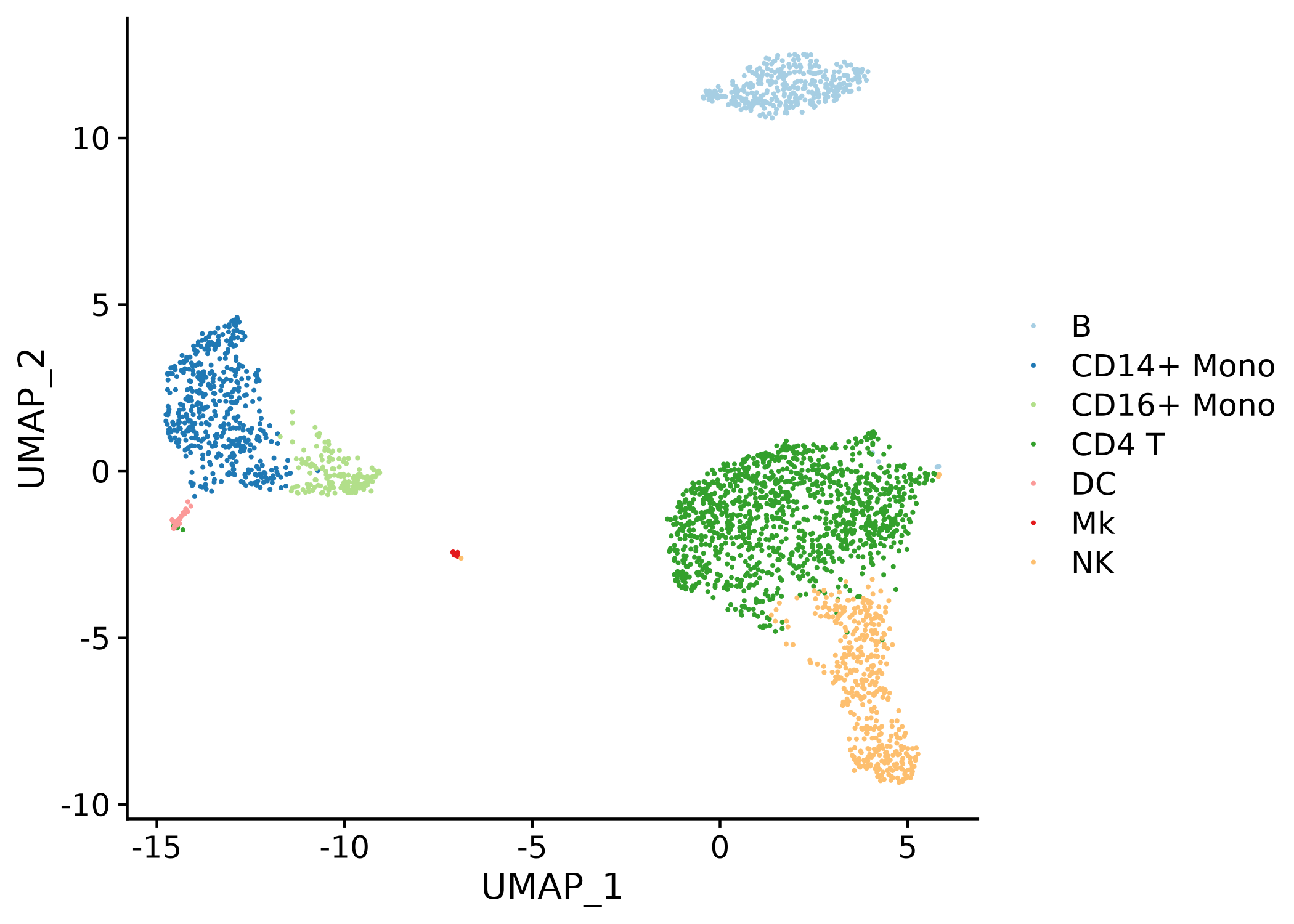
# plot assignments on a projection
plot_best_call(
cor_mat = res,
metadata = pbmc_meta,
cluster_col = "classified"
)
clustify() can take a clustered SingleCellExperiment or seurat object (from v2 up to v5) and assign identities.
# for SingleCellExperiment
sce_small <- sce_pbmc()
clustify(
input = sce_small, # an SCE object
ref_mat = cbmc_ref, # matrix of RNA-seq expression data for each cell type
cluster_col = "cell_type", # name of column in meta.data containing cell clusters
obj_out = TRUE # output SCE object with cell type inserted as "type" column
)
#> class: SingleCellExperiment
#> dim: 2000 2638
#> metadata(0):
#> assays(2): counts logcounts
#> rownames(2000): PPBP LYZ ... CLIC2 HEMGN
#> rowData names(0):
#> colnames(2638): AAACATACAACCAC AAACATTGAGCTAC ... TTTGCATGAGAGGC
#> TTTGCATGCCTCAC
#> colData names(8): cell_source sum ... type r
#> reducedDimNames(1): UMAP
#> mainExpName: NULL
#> altExpNames(0):
# for Seurat
library(Seurat)
s_small <- so_pbmc()
clustify(
input = s_small,
cluster_col = "RNA_snn_res.0.5",
ref_mat = cbmc_ref,
seurat_out = TRUE
)
#> An object of class Seurat
#> 2000 features across 2638 samples within 1 assay
#> Active assay: RNA (2000 features, 2000 variable features)
#> 2 layers present: counts, data
#> 1 dimensional reduction calculated: umap
# New output option, directly as a vector (in the order of the metadata), which can then be inserted into metadata dataframes and other workflows
clustify(
input = s_small,
cluster_col = "RNA_snn_res.0.5",
ref_mat = cbmc_ref,
vec_out = TRUE
)[1:10]
#> [1] "CD4 T" "B" "CD4 T" "CD14+ Mono" "NK"
#> [6] "CD4 T" "NK" "NK" "CD4 T" "CD16+ Mono"New reference matrix can be made directly from SingleCellExperiment and Seurat objects as well. Other scRNAseq experiment object types are supported as well.
# make reference from SingleCellExperiment objects
sce_small <- sce_pbmc()
sce_ref <- object_ref(
input = sce_small, # SCE object
cluster_col = "cell_type" # name of column in colData containing cell identities
)
# make reference from seurat objects
s_small <- so_pbmc()
s_ref <- seurat_ref(
seurat_object = s_small,
cluster_col = "RNA_snn_res.0.5"
)
head(s_ref)
#> 0 1 2 3 4 5
#> PPBP 0.04883837 0.06494743 0.28763857 0.09375021 0.35662599 0.2442300
#> LYZ 1.40165143 1.39466552 5.21550849 1.42699419 1.35146753 3.4034309
#> S100A9 0.55679700 0.58080250 4.91453355 0.62123058 0.58823794 2.6277996
#> IGLL5 0.03116080 0.04826212 0.02434753 2.44576997 0.03284986 0.2581198
#> GNLY 0.46041901 0.41001072 0.53592906 0.37877736 2.53161887 0.2903092
#> FTL 3.35611600 3.31062958 5.86217774 3.66698837 3.37056910 5.9518479
#> 6 7 8
#> PPBP 0.00000000 0.06527347 6.0941782
#> LYZ 1.32701580 4.84714962 2.5303912
#> S100A9 0.52098541 2.53310734 1.6775692
#> IGLL5 0.05247669 0.10986617 0.2501642
#> GNLY 4.70481754 0.46959958 0.3845813
#> FTL 3.38471536 4.21848878 4.5508242clustify_lists() handles identity assignment of matrix or SingleCellExperiment and seurat objects based on marker gene lists.
clustify_lists(
input = pbmc_matrix_small,
metadata = pbmc_meta,
cluster_col = "classified",
marker = pbmc_markers,
marker_inmatrix = FALSE
)
#> 0 1 2 3 4 5 6
#> Naive CD4 T 1.5639055 20.19469 31.77095 8.664074 23.844992 19.06931 19.06931
#> Memory CD4 T 1.5639055 20.19469 31.77095 10.568007 23.844992 17.97875 19.06931
#> CD14+ Mono 0.9575077 14.70716 76.21353 17.899569 11.687739 49.86699 16.83210
#> B 0.6564777 12.70976 31.77095 26.422929 13.536295 20.19469 13.53630
#> CD8 T 1.0785353 17.97875 31.82210 12.584823 31.822099 22.71234 40.45383
#> FCGR3A+ Mono 0.6564777 13.63321 72.43684 17.899569 9.726346 56.48245 14.61025
#> NK 0.6564777 14.61025 31.82210 7.757206 31.822099 22.71234 45.05072
#> DC 0.6564777 15.80598 63.34978 19.069308 13.758144 40.56298 17.97875
#> Platelet 0.5428889 13.34769 59.94938 14.215244 15.158755 46.92861 19.49246
#> 7 8
#> Naive CD4 T 6.165348 0.6055118
#> Memory CD4 T 6.165348 0.9575077
#> CD14+ Mono 25.181595 1.0785353
#> B 17.899569 0.1401901
#> CD8 T 7.882145 0.3309153
#> FCGR3A+ Mono 21.409177 0.3309153
#> NK 5.358651 0.3309153
#> DC 45.101877 0.1401901
#> Platelet 19.492465 59.9493793
clustify_lists(
input = s_small,
marker = pbmc_markers,
marker_inmatrix = FALSE,
cluster_col = "RNA_snn_res.0.5",
seurat_out = TRUE
)
#> An object of class Seurat
#> 2000 features across 2638 samples within 1 assay
#> Active assay: RNA (2000 features, 2000 variable features)
#> 2 layers present: counts, data
#> 1 dimensional reduction calculated: umapAdditional resources
Script for benchmarking, compatible with
scRNAseq_BenchmarkAdditional reference data (including tabula muris, immgen, etc) are available in a supplemental package
clustifyrdatahub. Also see list for individual downloads.See the FAQ for more details.