Plotting tSNE and UMAP results
clustifyr provides several functions to plot tSNE or UMAP results. The plot_dims() function will plot tSNE or UMAP data using a meta.data table and can color the cells based on cluster identity.
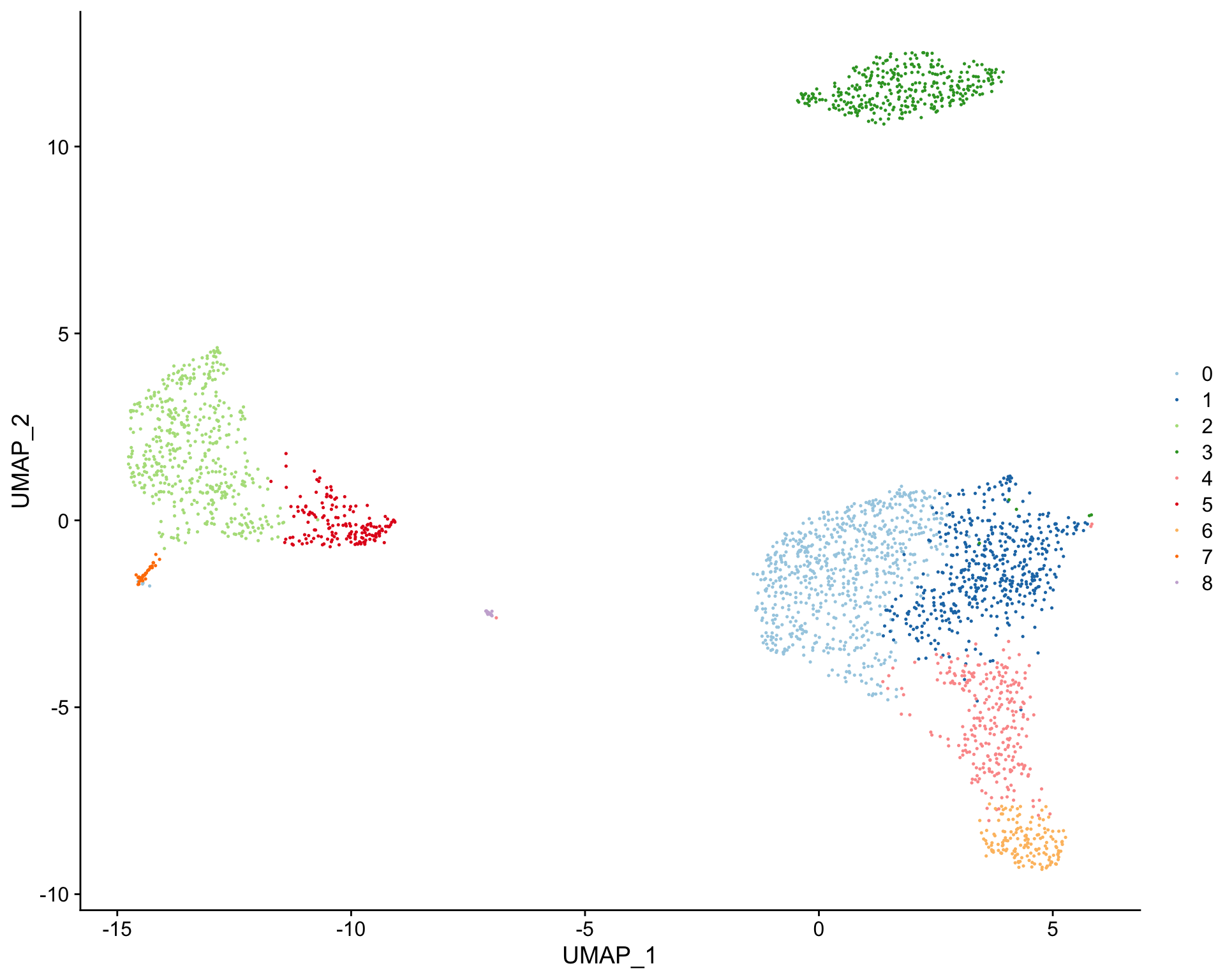
library(clustifyr) library(clustifyrdata) library(dplyr) library(tibble) # Matrix of normalized single-cell RNA-seq counts pbmc_matrix[1:3, 1:3] #> 3 x 3 sparse Matrix of class "dgCMatrix" #> AAACATACAACCAC AAACATTGAGCTAC AAACATTGATCAGC #> AL627309.1 . . . #> AP006222.2 . . . #> RP11-206L10.2 . . . # meta.data table containing cluster assignments for each cell pbmc_meta[1:5, ] #> orig.ident nCount_RNA nFeature_RNA percent.mt RNA_snn_res.0.5 #> AAACATACAACCAC pbmc3k 2419 779 3.0177759 1 #> AAACATTGAGCTAC pbmc3k 4903 1352 3.7935958 3 #> AAACATTGATCAGC pbmc3k 3147 1129 0.8897363 1 #> AAACCGTGCTTCCG pbmc3k 2639 960 1.7430845 2 #> AAACCGTGTATGCG pbmc3k 980 521 1.2244898 6 #> seurat_clusters classified UMAP_1 UMAP_2 #> AAACATACAACCAC 1 Memory CD4 T 2.341260 -3.0761487 #> AAACATTGAGCTAC 3 B 3.712070 11.7883301 #> AAACATTGATCAGC 1 Memory CD4 T 5.065264 -0.7839952 #> AAACCGTGCTTCCG 2 CD14+ Mono -11.783583 0.8343185 #> AAACCGTGTATGCG 6 NK 3.576498 -8.9778881 # Create tSNE and color cells based on cluster ID plot_dims( x = "UMAP_1", # name of column in the meta.data containing the data to plot on x-axis y = "UMAP_2", # name of column in the meta.data containing the data to plot on y-axis data = pbmc_meta, # meta.data table containing cluster assignments for each cell feature = "seurat_clusters" # name of column in meta.data to color cells by )
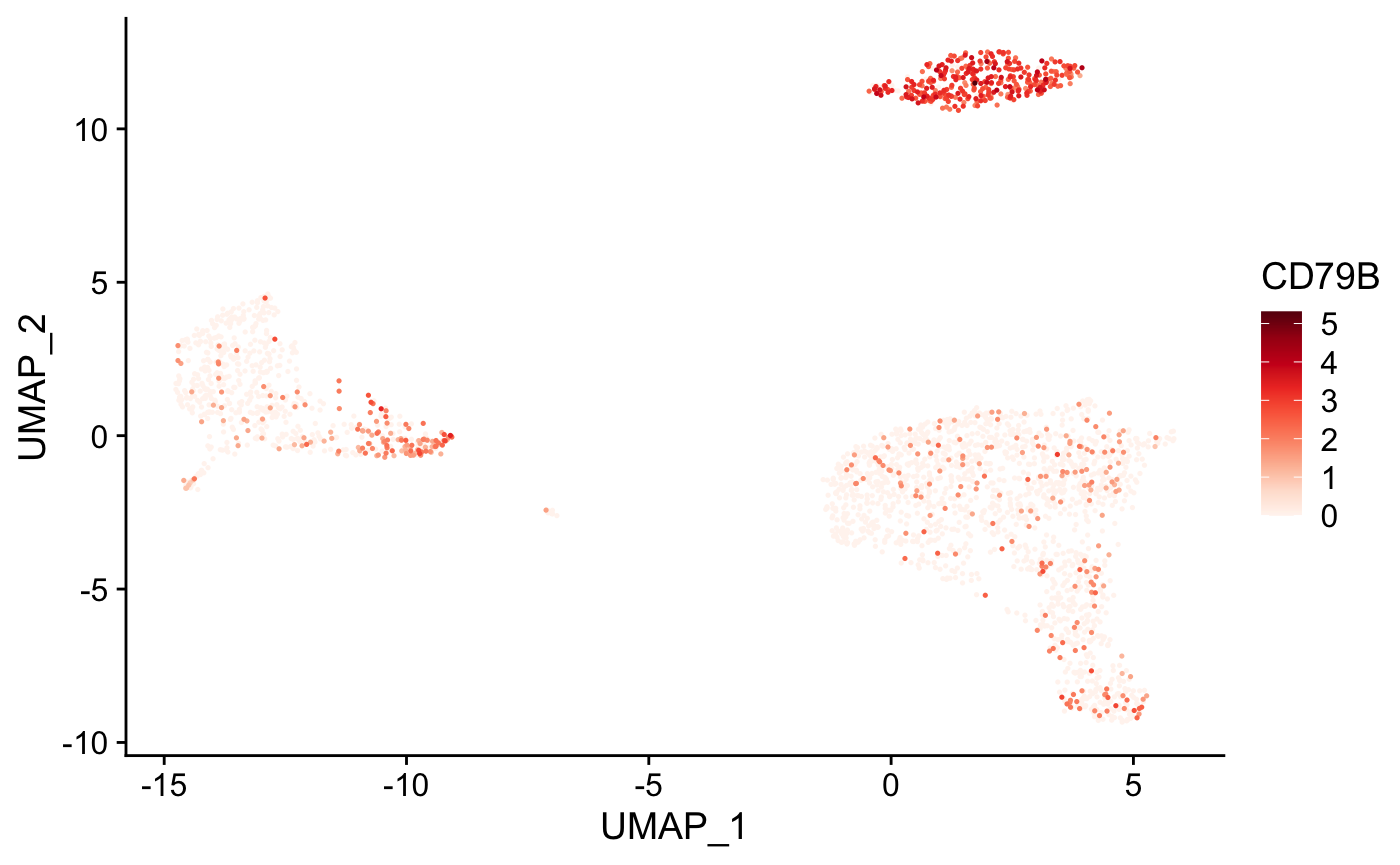
Cells can also be colored based on the expression level of a gene or list of genes using the plot_gene() function.
# Create tSNE and color cells based on gene expression plot_gene( x = "UMAP_1", # name of column in the meta.data containing the data to plot on x-axis y = "UMAP_2", # name of column in the meta.data containing the data to plot on y-axis expr_mat = pbmc_matrix, # matrix of normalized single-cell RNA-seq counts metadata = pbmc_meta %>% rownames_to_column("rn"), # meta.data table containing cluster assignments for each cell genes = c("CD79B", "CD8A"), # vector of gene names to color cells cell_col = "rn" # name of column in meta.data containing the cell IDs ) #> [[1]]
#>
#> [[2]]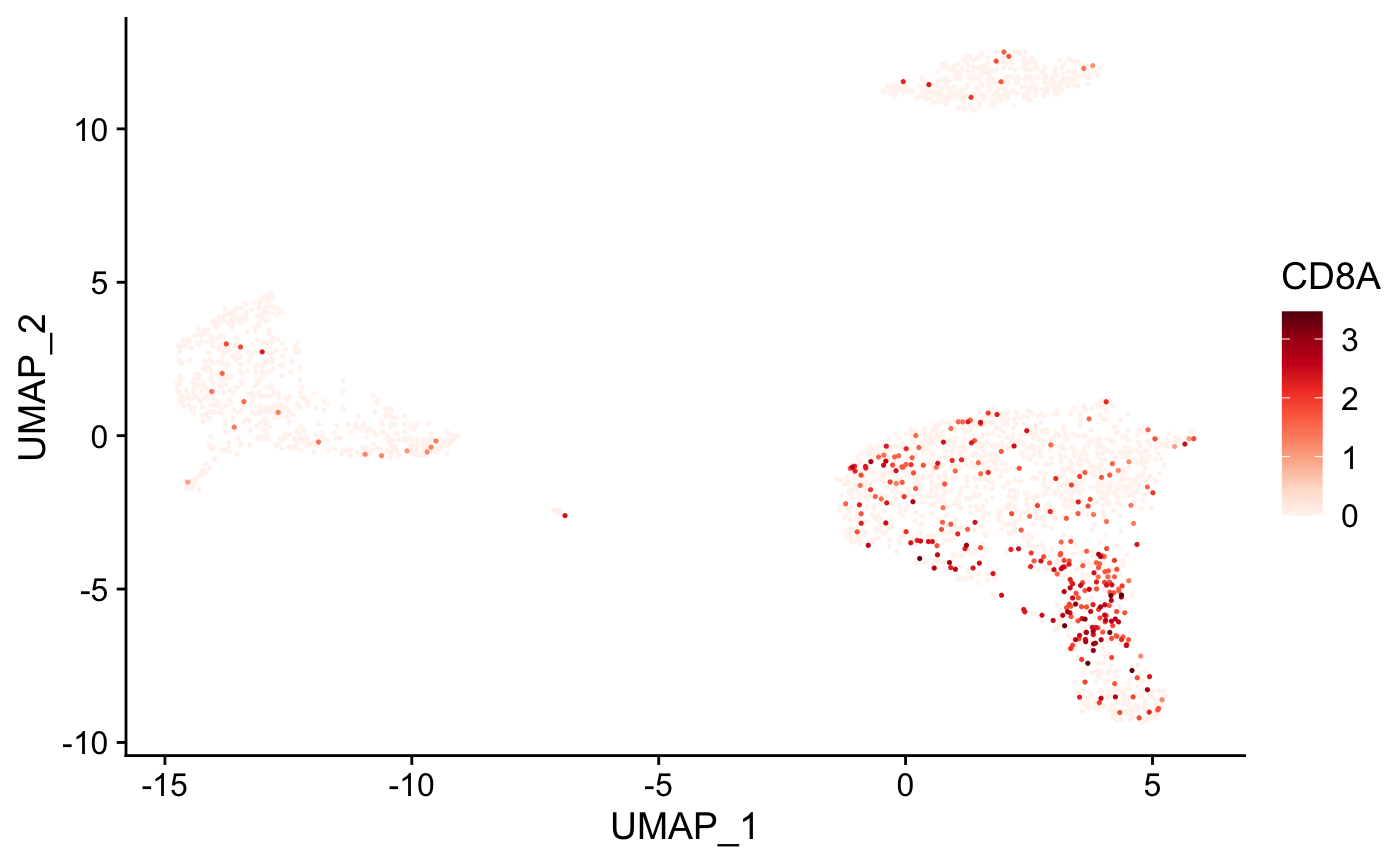
Visualizing clustifyr() results
The clustifyr() function outputs a matrix of correlation coefficients and clustify_lists() and clustify_nudge() output positive scores. clustifyr provides built-in functions to help visualize these results.
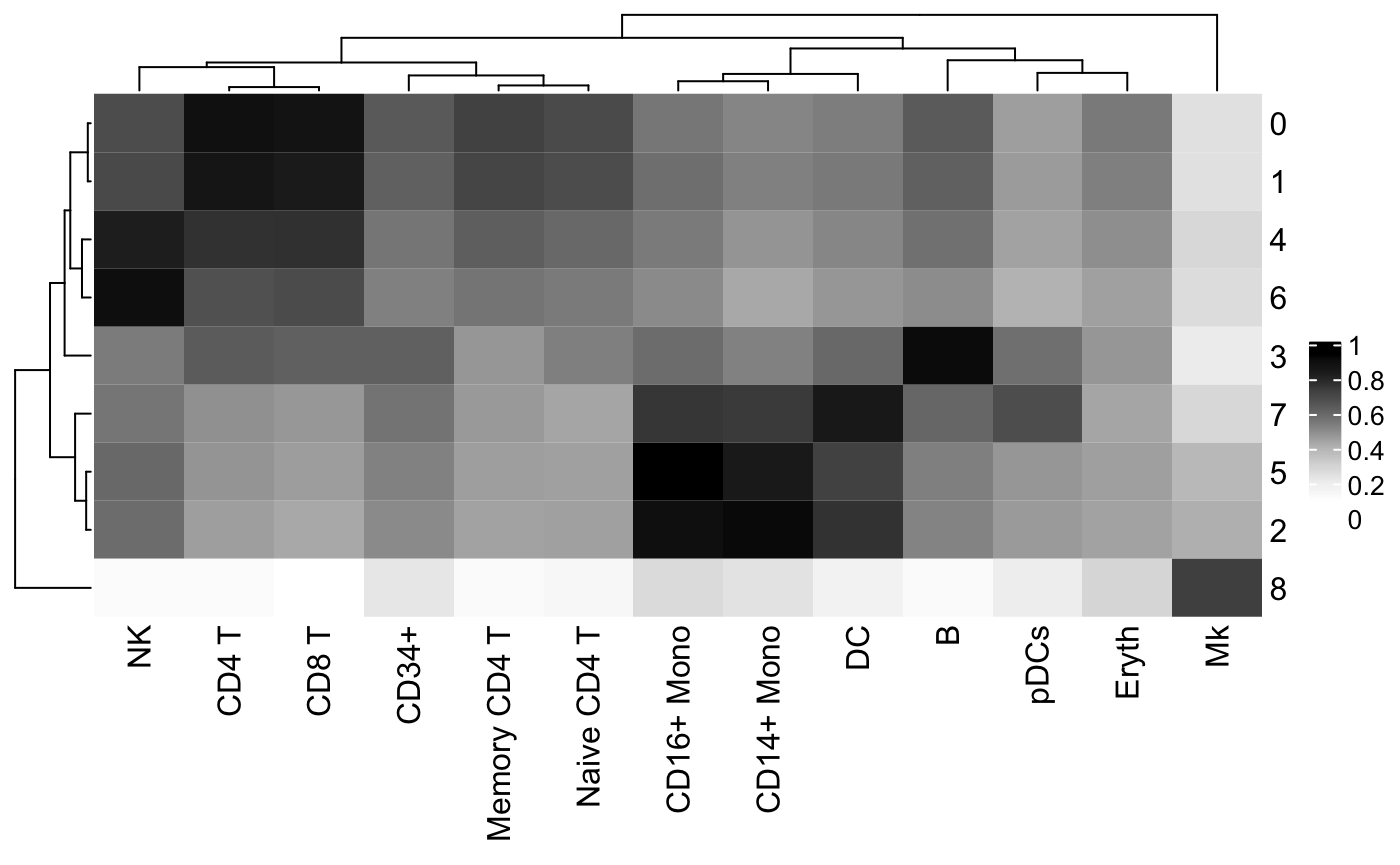
Cell type assignments can be assessed by plotting the clustifyr() correlation matrix as a heatmap using the plot_cor_heatmap() function.
# Run clustifyr() res <- clustify( input = pbmc_matrix, # matrix of normalized single-cell RNA-seq counts metadata = pbmc_meta, # meta.data table containing cluster assignments for each cell ref_mat = cbmc_ref, # reference matrix containing bulk RNA-seq data for each cell type query_genes = pbmc_vargenes, # list of highly varible genes identified with Seurat cluster_col = "seurat_clusters" # name of column in meta.data containing cell clusters ) #> [1] "use" # Create heatmap using the clustifyr correlation matrix plot_cor_heatmap( cor_mat = res # matrix of correlation coefficients from clustifyr() )
The plot_cor() function can also be used to create a tSNE for each cell type of interest and color the cells based on the correlation coefficients.
# Create a tSNE for each cell type of interest and color cells based on correlation coefficients plot_cor( x = "UMAP_1", # name of column in the meta.data containing the data to plot on x-axis y = "UMAP_2", # name of column in the meta.data containing the data to plot on y-axis cor_mat = res, # matrix of correlation coefficients from clustifyr() metadata = pbmc_meta, # meta.data table containing cluster assignments for each cell data_to_plot = colnames(res)[1:2], # name of cell type(s) to plot correlation coefficients cluster_col = "seurat_clusters" # name of column in meta.data containing cell clusters ) #> [[1]]
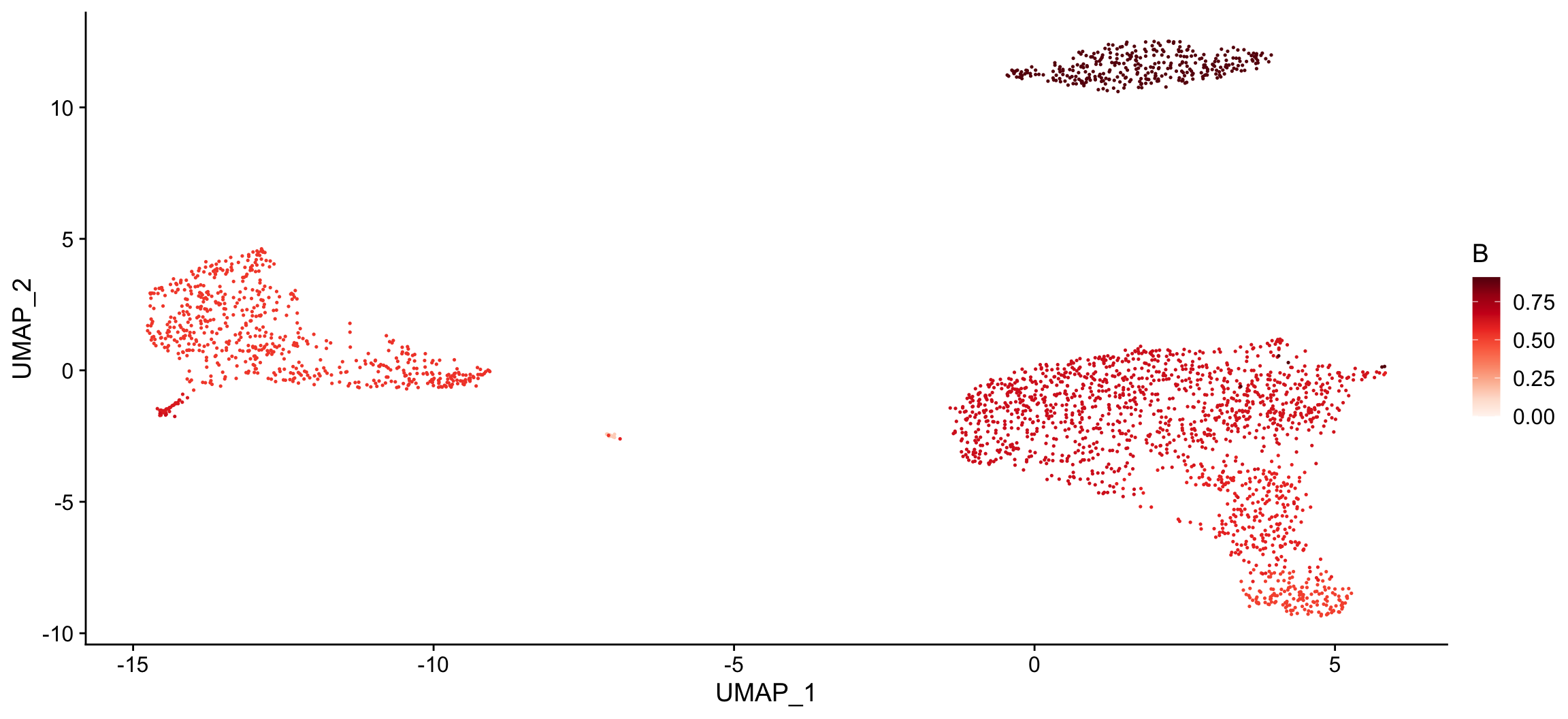
#>
#> [[2]]
Cell clusters can also be labeled using the plot_best_call() function, which takes the correlation matrix and labels cell clusters with the cell type that has the highest correlation coefficient.
# Create tSNE and label clusters with the cell type that has the highest correlation coefficient plot_best_call( cor_mat = res, # matrix of correlation coefficients from clustifyr() metadata = pbmc_meta, # meta.data table containing UMAP or tSNE data do_label = TRUE, # should the feature label be shown on each cluster? do_legend = FALSE, # should the legend be shown? cluster_col = "seurat_clusters" )
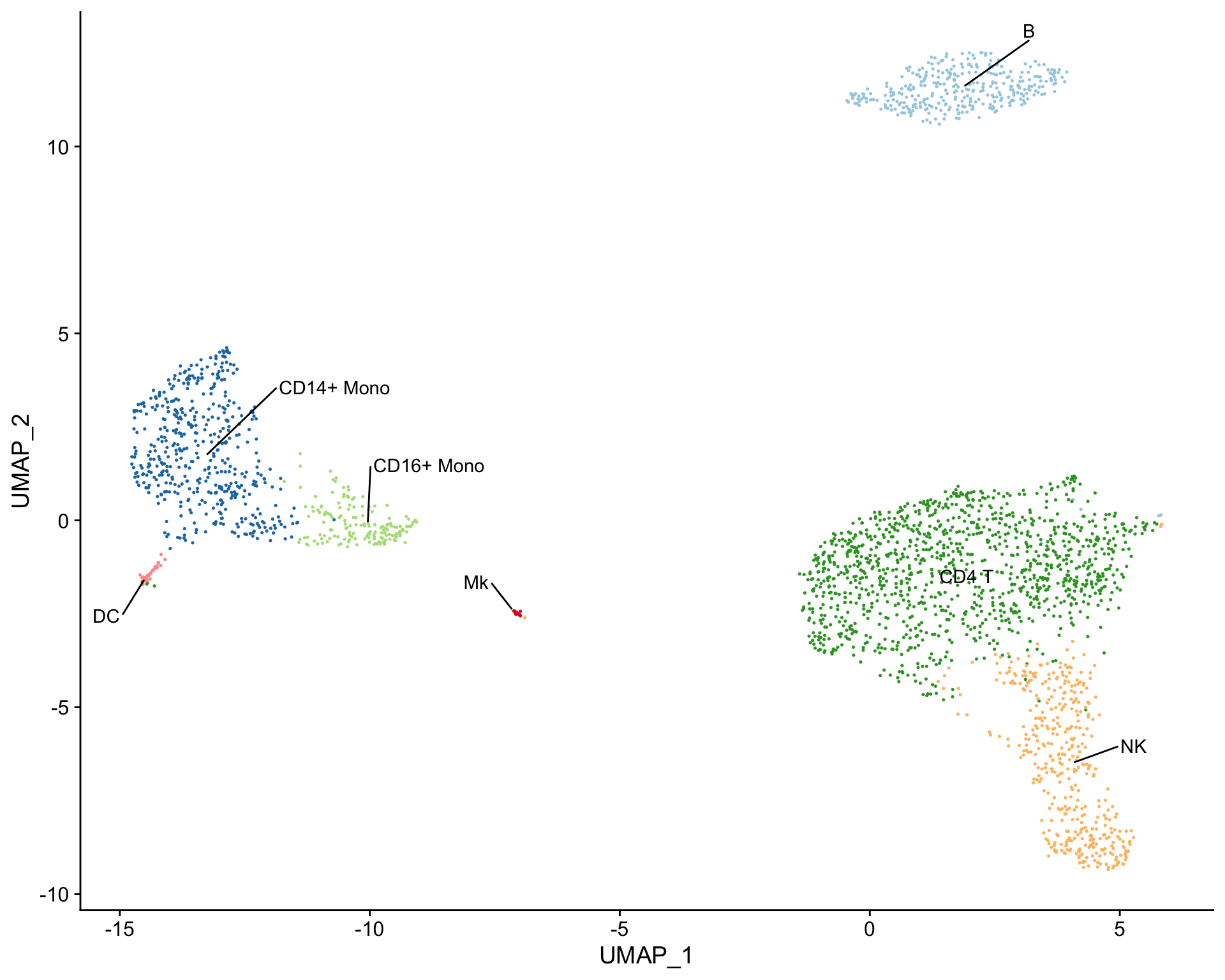
Assessing clustifyr() accuracy
The clustifyr() results can also be evaluated by over-clustering the data and comparing the cell type assignments before and after over-clustering. This is accomplished using the overcluster_test() function. The cell type assignments should be similar with and without over-clustering.
# Overcluster cells and compare cell type assignments with and without over-clustering overcluster_test( expr = pbmc_matrix, # matrix of normalized single-cell RNA-seq counts metadata = pbmc_meta, # meta.data table containing UMAP or tSNE data ref_mat = cbmc_ref, # reference matrix containing bulk RNA-seq data for each cell type cluster_col = "seurat_clusters", # name of column in meta.data containing cell clusters n = 5 # expand cluster number n-fold for overclustering ) #> [1] "use" #> [1] "use"
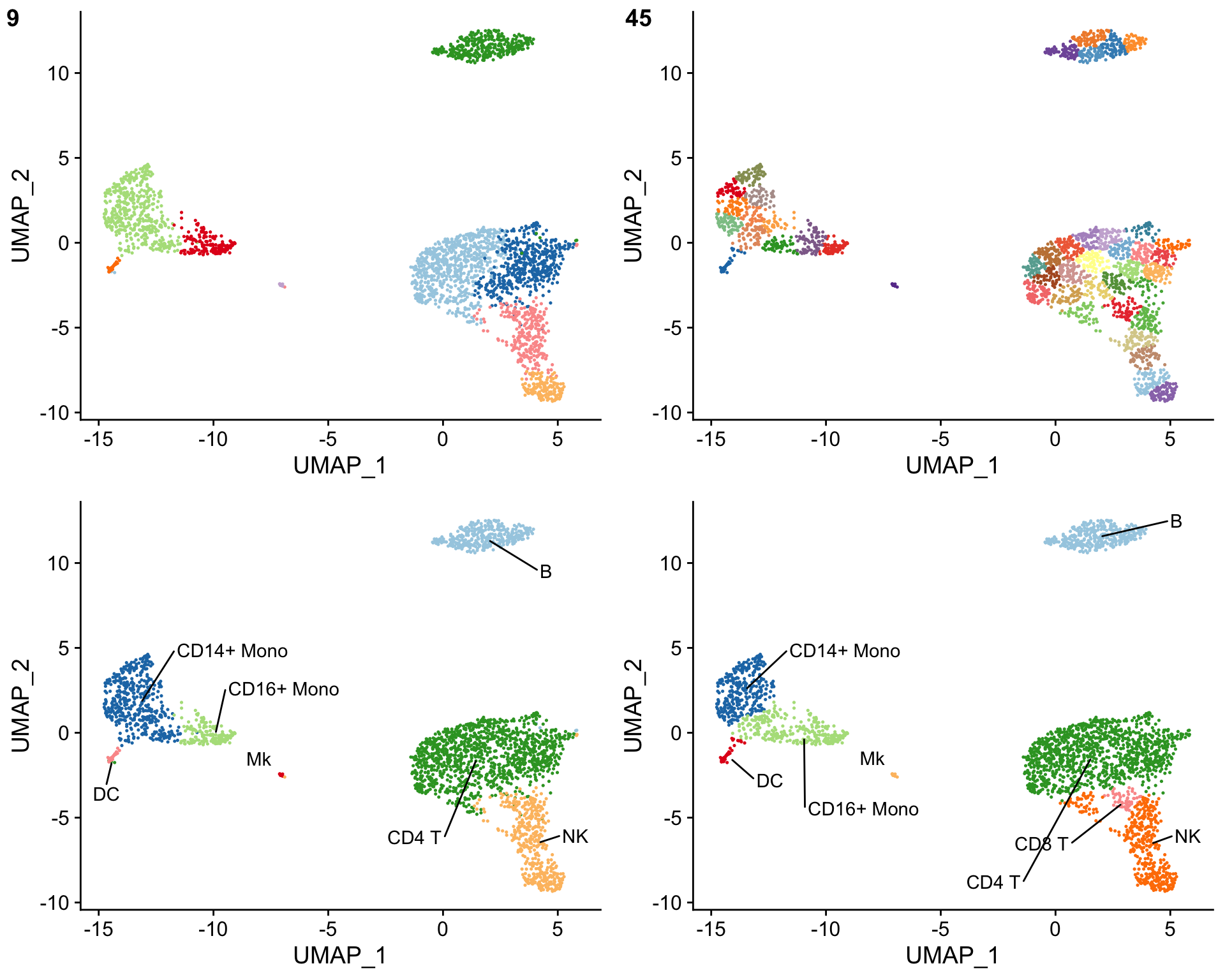
The cell types from the bulk RNA-seq reference matrix can also be mixed together using the make_comb_ref() function to assess the specificity of the cell type assignments. If a cluster shows a higher correlation when using the mixed reference matrix, this suggests that the cluster contains multiple cell types.
# Create reference containing different combinations of the bulk RNA-seq data comb_ref <- make_comb_ref( ref_mat = cbmc_ref # reference matrix containing bulk RNA-seq data for each cell type ) # Peek at the new combined reference comb_ref[1:5, 1:5] # Run clustifyr() using the combined reference comb_res <- clustify( input = pbmc_matrix, # matrix of normalized single-cell RNA-seq counts metadata = pbmc_meta, # meta.data table containing cluster assignments for each cell ref_mat = comb_ref, # reference matrix containing bulk RNA-seq data for each cell type query_genes = pbmc_vargenes, # list of highly varible genes identified with Seurat cluster_col = "seurat_clusters" # name of column in meta.data containing cell clusters ) # Create tSNE and label clusters with the assigned cell types from the combined reference plot_best_call( cor_mat = comb_res, # matrix of correlation coefficients from clustifyr() metadata = pbmc_meta, # meta.data table containing UMAP or tSNE data do_label = TRUE, # should the feature label be shown on each cluster? do_legend = FALSE, # should the legend be shown? cluster_col = "seurat_clusters" )
Plotting other attributes from the meta.data table
Visualization of other attributes shared in the metadata between ref and query by plot_cols, such as nGene, nUMI, mt_percentage, as another way of identity confirmation after clustify. Certain cell types have distinct patterns, more genes detected, for example.