Operations can be performed on merged intervals by specifying name-value
pairs. Default max_dist of 0 means book-ended intervals are
merged.
Arguments
Details
input tbls are grouped by chrom by default, and additional
groups can be added using dplyr::group_by(). For example,
grouping by strand will constrain analyses to the same strand. To
compare opposing strands across two tbls, strands on the y tbl can
first be inverted using flip_strands().
See also
https://bedtools.readthedocs.io/en/latest/content/tools/merge.html
Other single set operations:
bed_cluster(),
bed_complement(),
bed_flank(),
bed_genomecov(),
bed_partition(),
bed_shift(),
bed_slop()
Examples
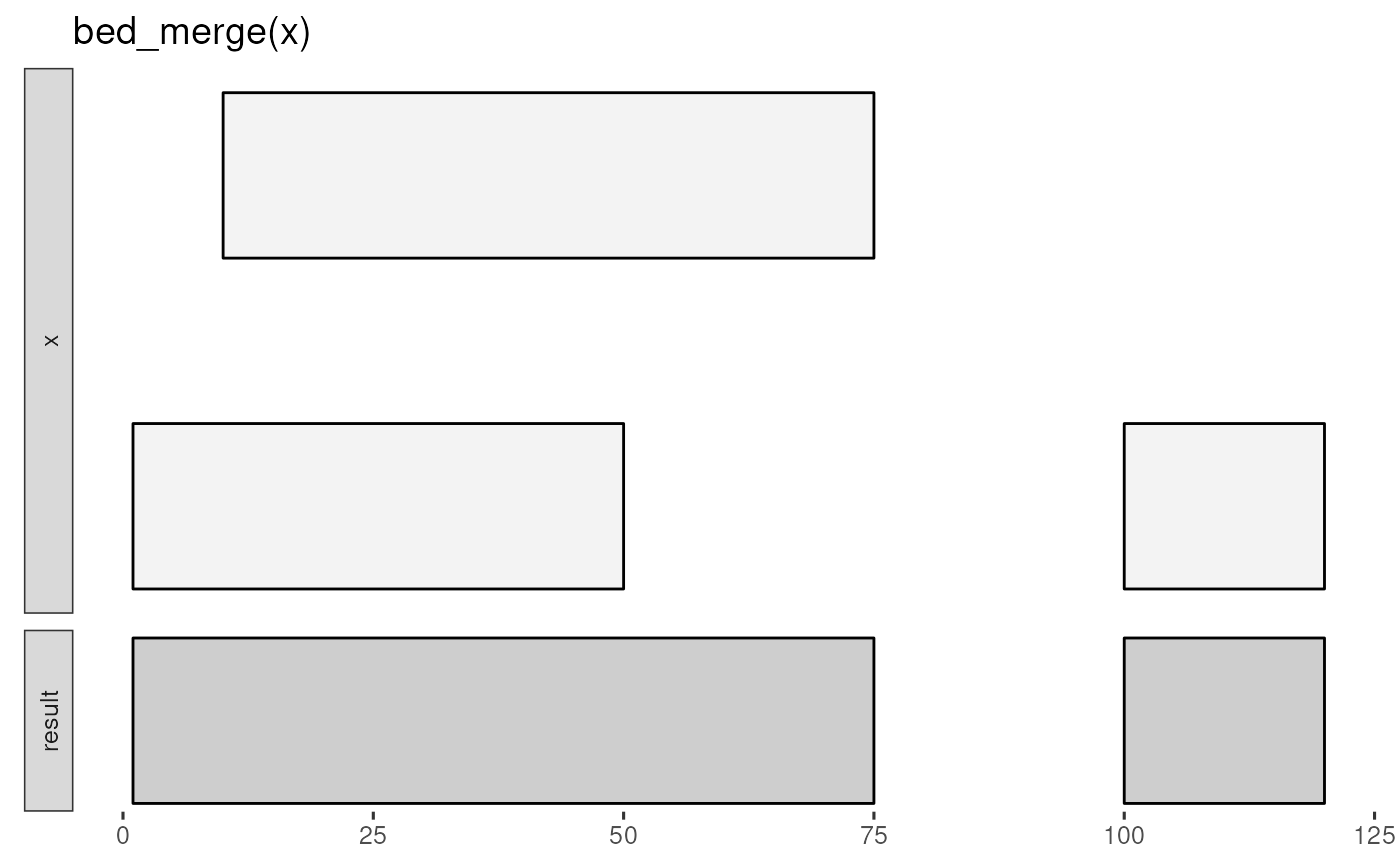
x <- tibble::tribble(
~chrom, ~start, ~end,
"chr1", 1, 50,
"chr1", 10, 75,
"chr1", 100, 120
)
bed_glyph(bed_merge(x))
 x <- tibble::tribble(
~chrom, ~start, ~end, ~value, ~strand,
"chr1", 1, 50, 1, "+",
"chr1", 100, 200, 2, "+",
"chr1", 150, 250, 3, "-",
"chr2", 1, 25, 4, "+",
"chr2", 200, 400, 5, "-",
"chr2", 400, 500, 6, "+",
"chr2", 450, 550, 7, "+"
)
bed_merge(x)
#> # A tibble: 4 × 3
#> chrom start end
#> <chr> <dbl> <dbl>
#> 1 chr1 1 50
#> 2 chr1 100 250
#> 3 chr2 1 25
#> 4 chr2 200 550
bed_merge(x, max_dist = 100)
#> # A tibble: 3 × 3
#> chrom start end
#> <chr> <dbl> <dbl>
#> 1 chr1 1 250
#> 2 chr2 1 25
#> 3 chr2 200 550
# merge intervals on same strand
bed_merge(dplyr::group_by(x, strand))
#> # A tibble: 6 × 4
#> # Groups: strand [2]
#> chrom start end strand
#> <chr> <dbl> <dbl> <chr>
#> 1 chr1 1 50 +
#> 2 chr1 100 200 +
#> 3 chr1 150 250 -
#> 4 chr2 1 25 +
#> 5 chr2 400 550 +
#> 6 chr2 200 400 -
bed_merge(x, .value = sum(value))
#> # A tibble: 4 × 4
#> chrom start end .value
#> <chr> <dbl> <dbl> <dbl>
#> 1 chr1 1 50 1
#> 2 chr1 100 250 5
#> 3 chr2 1 25 4
#> 4 chr2 200 550 18
x <- tibble::tribble(
~chrom, ~start, ~end, ~value, ~strand,
"chr1", 1, 50, 1, "+",
"chr1", 100, 200, 2, "+",
"chr1", 150, 250, 3, "-",
"chr2", 1, 25, 4, "+",
"chr2", 200, 400, 5, "-",
"chr2", 400, 500, 6, "+",
"chr2", 450, 550, 7, "+"
)
bed_merge(x)
#> # A tibble: 4 × 3
#> chrom start end
#> <chr> <dbl> <dbl>
#> 1 chr1 1 50
#> 2 chr1 100 250
#> 3 chr2 1 25
#> 4 chr2 200 550
bed_merge(x, max_dist = 100)
#> # A tibble: 3 × 3
#> chrom start end
#> <chr> <dbl> <dbl>
#> 1 chr1 1 250
#> 2 chr2 1 25
#> 3 chr2 200 550
# merge intervals on same strand
bed_merge(dplyr::group_by(x, strand))
#> # A tibble: 6 × 4
#> # Groups: strand [2]
#> chrom start end strand
#> <chr> <dbl> <dbl> <chr>
#> 1 chr1 1 50 +
#> 2 chr1 100 200 +
#> 3 chr1 150 250 -
#> 4 chr2 1 25 +
#> 5 chr2 400 550 +
#> 6 chr2 200 400 -
bed_merge(x, .value = sum(value))
#> # A tibble: 4 × 4
#> chrom start end .value
#> <chr> <dbl> <dbl> <dbl>
#> 1 chr1 1 50 1
#> 2 chr1 100 250 5
#> 3 chr2 1 25 4
#> 4 chr2 200 550 18