Familiar tools, natively in R
The functions in valr have similar names to their
BEDtools counterparts, and so will be familiar to users
coming from the BEDtools suite. Similar to pybedtools,
valr has a terse syntax:
library(valr)
library(dplyr)
snps <- read_bed(valr_example("hg19.snps147.chr22.bed.gz"))
genes <- read_bed(valr_example("genes.hg19.chr22.bed.gz"))
# find snps in intergenic regions
intergenic <- bed_subtract(snps, genes)
# distance from intergenic snps to nearest gene
nearby <- bed_closest(intergenic, genes)
nearby |>
select(starts_with("name"), .overlap, .dist) |>
filter(abs(.dist) < 1000)
#> # A tibble: 555 × 4
#> name.x name.y .overlap .dist
#> <chr> <chr> <int> <int>
#> 1 rs2261631 P704P 0 -268
#> 2 rs570770556 POTEH 0 -913
#> 3 rs538163832 POTEH 0 -953
#> 4 rs9606135 TPTEP1 0 -422
#> 5 rs11912392 ANKRD62P1-PARP4P3 0 105
#> 6 rs555445266 ANKRD62P1-PARP4P3 0 1
#> 7 rs8136454 BC038197 0 356
#> 8 rs576739562 XKR3 0 1
#> 9 rs5992556 XKR3 0 -456
#> 10 rs114101676 GAB4 0 474
#> # ℹ 545 more rowsInput data
valr assigns common column names to facilitate
comparisons between tbls. All tbls will have chrom,
start, and end columns, and some tbls from
multi-column formats will have additional pre-determined column names.
See the read_bed() documentation for details.
bed_file <- valr_example("3fields.bed.gz")
read_bed(bed_file) # accepts filepaths or URLs
#> # A tibble: 10 × 3
#> chrom start end
#> <chr> <dbl> <dbl>
#> 1 chr1 11873 14409
#> 2 chr1 14361 19759
#> 3 chr1 14406 29370
#> 4 chr1 34610 36081
#> 5 chr1 69090 70008
#> 6 chr1 134772 140566
#> 7 chr1 321083 321115
#> 8 chr1 321145 321207
#> 9 chr1 322036 326938
#> 10 chr1 327545 328439valr can also operate on BED-like data.frames already
constructed in R, provided that columns named chrom,
start and end are present. New tbls can also
be constructed as either tibbles or base R
data.frames.
bed <- tribble(
~chrom , ~start , ~end ,
"chr1" , 1657492 , 2657492 ,
"chr2" , 2501324 , 3094650
)
bed
#> # A tibble: 2 × 3
#> chrom start end
#> <chr> <dbl> <dbl>
#> 1 chr1 1657492 2657492
#> 2 chr2 2501324 3094650bigWig and bigBed files
Signal (bigWig) and feature (bigBed) files are read with
read_bigwig() and read_bigbed(), which accept
local paths or http(s):// URLs.
bw <- valr_example("hg19.dnase1.bw")
read_bigwig(bw)
#> # A tibble: 2,587 × 4
#> chrom start end value
#> <chr> <int> <int> <dbl>
#> 1 chr22 16050284 16050464 1
#> 2 chr22 16051024 16051204 1
#> 3 chr22 16051224 16051764 1
#> 4 chr22 16052724 16052904 1
#> 5 chr22 16053224 16053404 1
#> 6 chr22 16054584 16054644 1
#> 7 chr22 16054644 16054764 2
#> 8 chr22 16054764 16054824 1
#> 9 chr22 16056404 16056424 1
#> 10 chr22 16056424 16056584 2
#> # ℹ 2,577 more rowsThe multiple-set operations (bed_intersect(),
bed_map(), bed_coverage(),
bed_subtract(), and bed_window()) also accept
a bigWig or bigBed path or URL directly in place of a y
tbl. Only the regions spanned by x are read from the file,
so large (and remote) files are queried without loading them in
full.
peaks <- tribble(
~chrom , ~start , ~end ,
"chr22", 16100000 , 16150000 ,
"chr22", 16500000 , 16550000
)
# sum DNase I signal over each peak, reading only those regions from the file
bed_map(peaks, bw, .signal = sum(value))
#> # A tibble: 2 × 4
#> chrom start end .signal
#> <chr> <dbl> <dbl> <dbl>
#> 1 chr22 16100000 16150000 101
#> 2 chr22 16500000 16550000 17Interval coordinates
valr adheres to the BED format which
specifies that the start position for an interval is zero based and the
end position is one-based. The first position in a chromosome is 0. The
end position for a chromosome is one position passed the last base, and
is not included in the interval. For example:
# a chromosome 100 basepairs in length
chrom <- tribble(
~chrom , ~start , ~end ,
"chr1" , 0 , 100
)
chrom
#> # A tibble: 1 × 3
#> chrom start end
#> <chr> <dbl> <dbl>
#> 1 chr1 0 100
# single base-pair intervals
bases <- tribble(
~chrom , ~start , ~end ,
"chr1" , 0 , 1 , # first base of chromosome
"chr1" , 1 , 2 , # second base of chromosome
"chr1" , 99 , 100 # last base of chromosome
)
bases
#> # A tibble: 3 × 3
#> chrom start end
#> <chr> <dbl> <dbl>
#> 1 chr1 0 1
#> 2 chr1 1 2
#> 3 chr1 99 100Remote databases
Remote databases can be accessed with db_ucsc() (to
access the UCSC Browser) and db_ensembl() (to access
Ensembl databases).
Visual documentation
The bed_glyph() tool illustrates the results of
operations in valr, similar to those found in the
BEDtools documentation. This glyph shows the result of
intersecting x and y intervals with
bed_intersect():
x <- tribble(
~chrom , ~start , ~end ,
"chr1" , 25 , 50 ,
"chr1" , 100 , 125
)
y <- tribble(
~chrom , ~start , ~end ,
"chr1" , 30 , 75
)
bed_glyph(bed_intersect(x, y))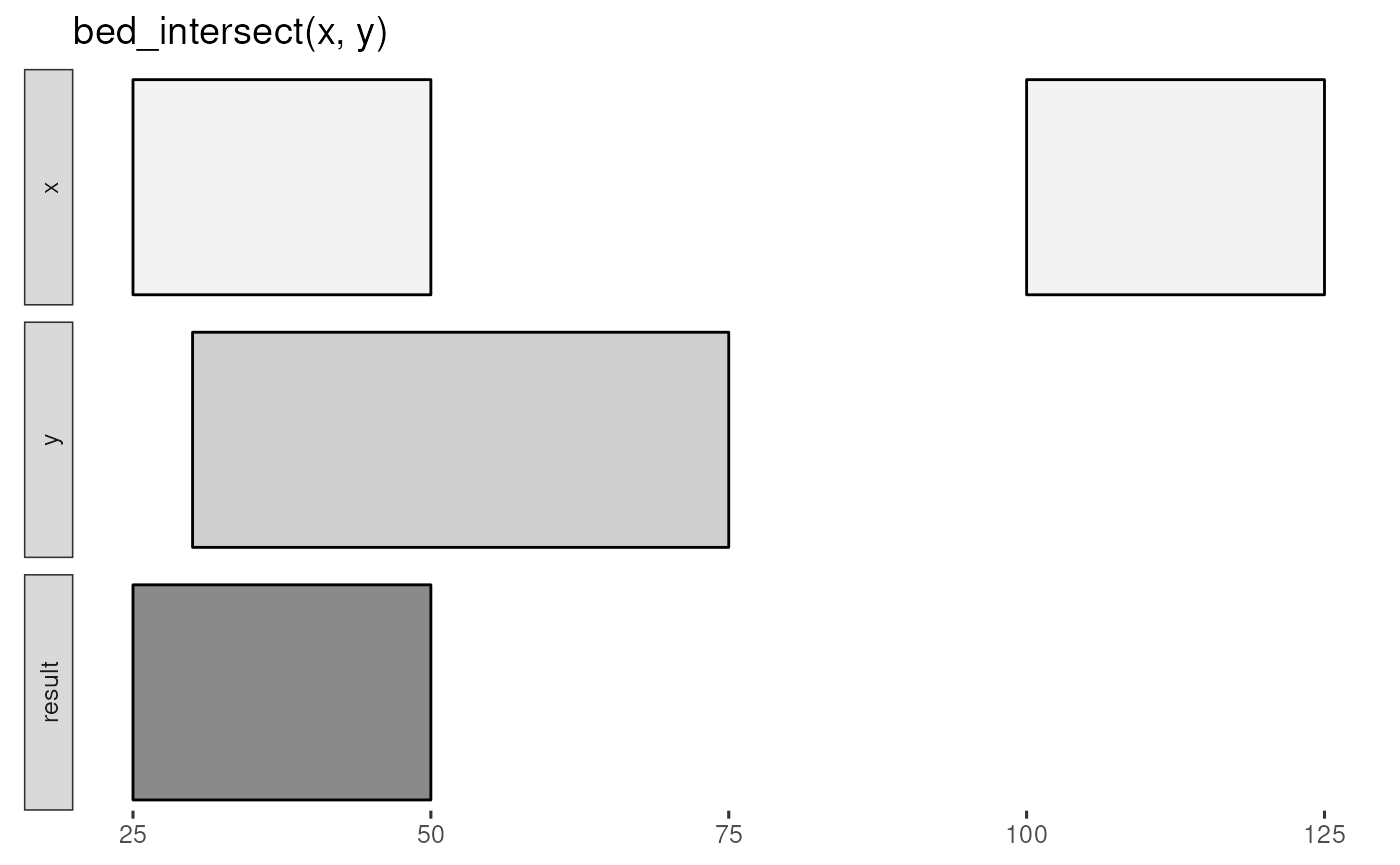
And this glyph illustrates bed_merge():
x <- tribble(
~chrom , ~start , ~end ,
"chr1" , 1 , 50 ,
"chr1" , 10 , 75 ,
"chr1" , 100 , 120
)
bed_glyph(bed_merge(x))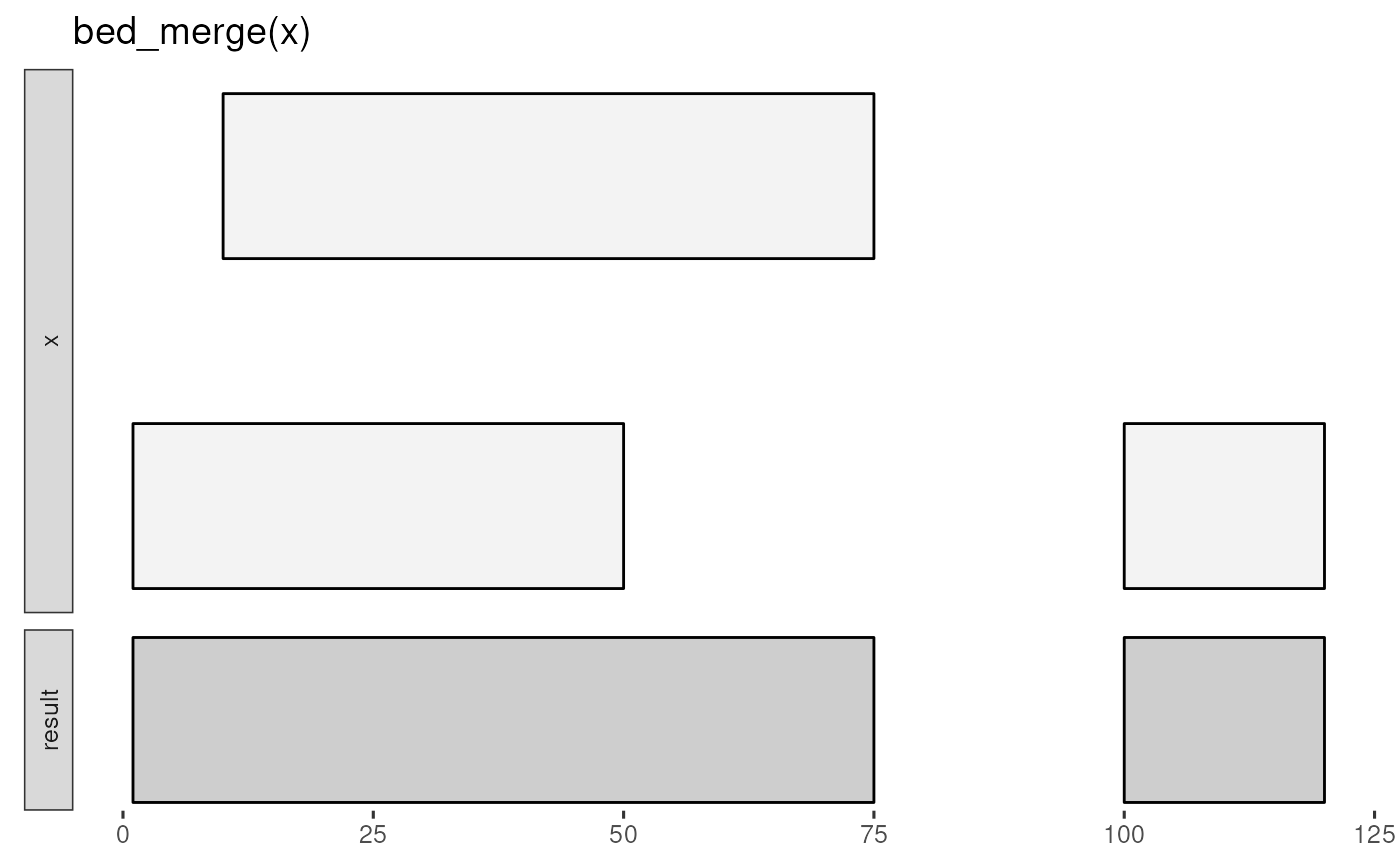
Grouping data
The group_by function in dplyr can be used to perform
functions on subsets of single and multiple data_frames.
Functions in valr leverage grouping to enable a variety of
comparisons. For example, intervals can be grouped by
strand to perform comparisons among intervals on the same
strand.
x <- tribble(
~chrom , ~start , ~end , ~strand ,
"chr1" , 1 , 100 , "+" ,
"chr1" , 50 , 150 , "+" ,
"chr2" , 100 , 200 , "-"
)
y <- tribble(
~chrom , ~start , ~end , ~strand ,
"chr1" , 50 , 125 , "+" ,
"chr1" , 50 , 150 , "-" ,
"chr2" , 50 , 150 , "+"
)
# intersect tbls by strand
x <- group_by(x, strand)
y <- group_by(y, strand)
bed_intersect(x, y)
#> # A tibble: 2 × 8
#> chrom start.x end.x strand.x start.y end.y strand.y .overlap
#> <chr> <dbl> <dbl> <chr> <dbl> <dbl> <chr> <int>
#> 1 chr1 1 100 + 50 125 + 50
#> 2 chr1 50 150 + 50 125 + 75Comparisons between intervals on opposite strands are done using the
flip_strands() function:
x <- group_by(x, strand)
y <- flip_strands(y)
y <- group_by(y, strand)
bed_intersect(x, y)
#> # A tibble: 3 × 8
#> chrom start.x end.x strand.x start.y end.y strand.y .overlap
#> <chr> <dbl> <dbl> <chr> <dbl> <dbl> <chr> <int>
#> 1 chr1 1 100 + 50 150 + 50
#> 2 chr1 50 150 + 50 150 + 100
#> 3 chr2 100 200 - 50 150 - 50Both single set (e.g. bed_merge()) and multi set
operations will respect groupings in the input intervals.
Column specification
Columns in BEDtools are referred to by position:
# calculate the mean of column 6 for intervals in `b` that overlap with `a`
bedtools map -a a.bed -b b.bed -c 6 -o meanIn valr, columns are referred to by name and can be used
in multiple name/value expressions for summaries.
Getting started
Meta-analysis
This demonstration illustrates how to use valr tools to
perform a “meta-analysis” of signals relative to genomic features. Here
we to analyze the distribution of histone marks surrounding
transcription start sites.
First we load libraries and relevant data.
# `valr_example()` identifies the path of example files
bedfile <- valr_example("genes.hg19.chr22.bed.gz")
genomefile <- valr_example("hg19.chrom.sizes.gz")
bgfile <- valr_example("hela.h3k4.chip.bg.gz")
genes <- read_bed(bedfile)
genome <- read_genome(genomefile)
y <- read_bedgraph(bgfile)Then we generate 1 bp intervals to represent transcription start
sites (TSSs). We focus on + strand genes, but
- genes are easily accommodated by filtering them and using
bed_makewindows() with reversed window
numbers.
# generate 1 bp TSS intervals, `+` strand only
tss <- genes |>
filter(strand == "+") |>
mutate(end = start + 1)
# 1000 bp up and downstream
region_size <- 1000
# 50 bp windows
win_size <- 50
# add slop to the TSS, break into windows and add a group
x <- tss |>
bed_slop(genome, both = region_size) |>
bed_makewindows(win_size)
x
#> # A tibble: 13,530 × 7
#> chrom start end name score strand .win_id
#> <chr> <dbl> <dbl> <chr> <chr> <chr> <int>
#> 1 chr22 16161065 16161115 LINC00516 3 + 1
#> 2 chr22 16161115 16161165 LINC00516 3 + 2
#> 3 chr22 16161165 16161215 LINC00516 3 + 3
#> 4 chr22 16161215 16161265 LINC00516 3 + 4
#> 5 chr22 16161265 16161315 LINC00516 3 + 5
#> 6 chr22 16161315 16161365 LINC00516 3 + 6
#> 7 chr22 16161365 16161415 LINC00516 3 + 7
#> 8 chr22 16161415 16161465 LINC00516 3 + 8
#> 9 chr22 16161465 16161515 LINC00516 3 + 9
#> 10 chr22 16161515 16161565 LINC00516 3 + 10
#> # ℹ 13,520 more rowsNow we use the .win_id group with bed_map()
to calculate a sum by mapping y signals onto the intervals
in x. These data are regrouped by .win_id and
a summary with mean and sd values is
calculated.
# map signals to TSS regions and calculate summary statistics.
res <- bed_map(x, y, win_sum = sum(value, na.rm = TRUE)) |>
group_by(.win_id) |>
summarize(
win_mean = mean(win_sum, na.rm = TRUE),
win_sd = sd(win_sum, na.rm = TRUE)
)
res
#> # A tibble: 41 × 3
#> .win_id win_mean win_sd
#> <int> <dbl> <dbl>
#> 1 1 101. 85.8
#> 2 2 111. 81.1
#> 3 3 123. 99.1
#> 4 4 116. 96.3
#> 5 5 116. 102.
#> 6 6 125. 95.1
#> 7 7 123. 94.4
#> 8 8 128. 91.5
#> 9 9 130. 95.7
#> 10 10 130. 88.8
#> # ℹ 31 more rowsFinally, these summary statistics are used to construct a plot that illustrates histone density surrounding TSSs.
x_labels <- seq(
-region_size,
region_size,
by = win_size * 5
)
x_breaks <- seq(1, 41, by = 5)
sd_limits <- aes(
ymax = win_mean + win_sd,
ymin = win_mean - win_sd
)
ggplot(
res,
aes(
x = .win_id,
y = win_mean
)
) +
geom_point() +
geom_pointrange(sd_limits) +
scale_x_continuous(
labels = x_labels,
breaks = x_breaks
) +
labs(
x = "Position (bp from TSS)",
y = "Signal",
title = "Human H3K4me3 signal near transcription start sites"
) +
theme_classic()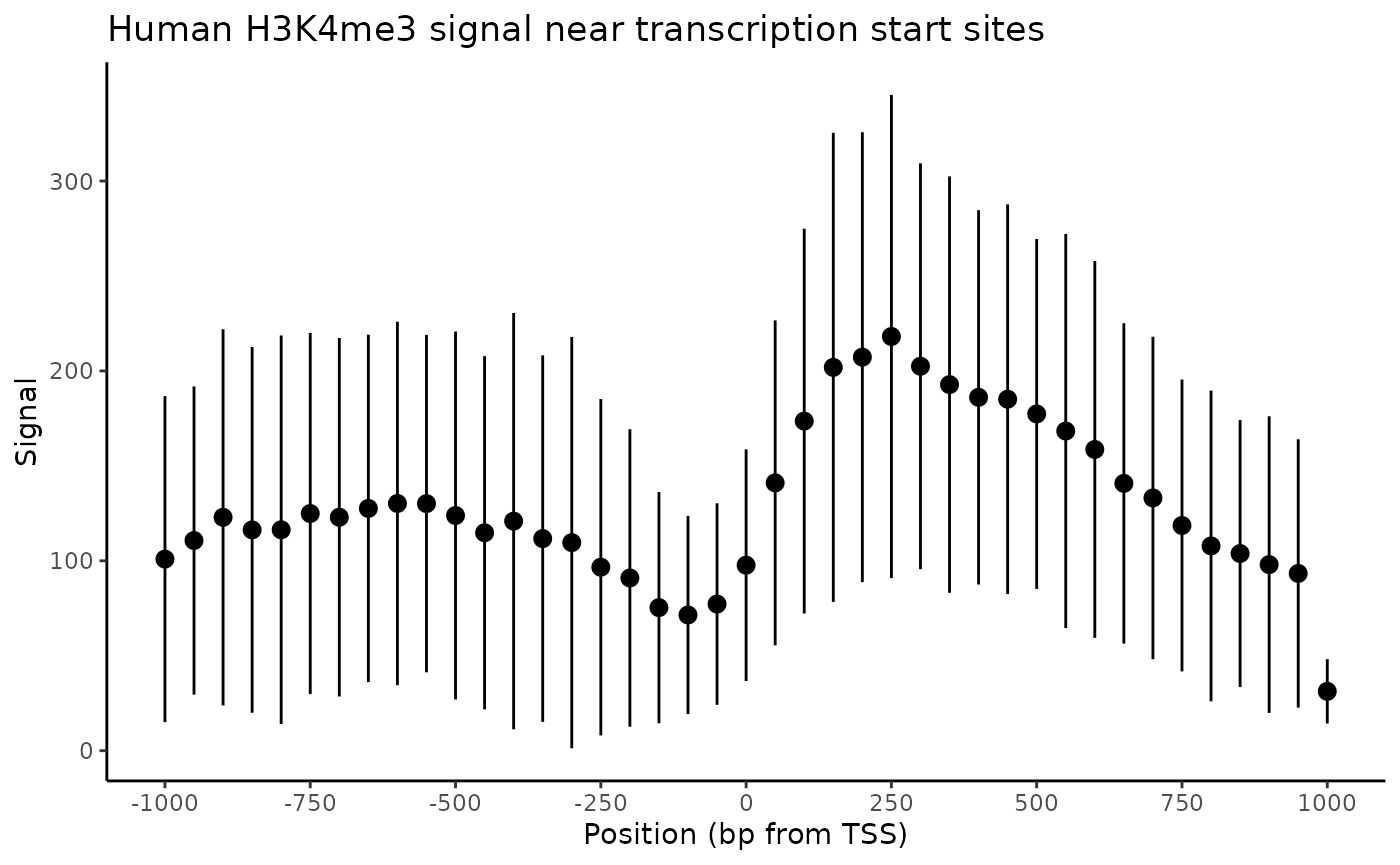
Related work
The Python library pybedtools wraps BEDtools.
The R packages GenomicRanges, bedr, IRanges and GenometriCorr provide similar capability with a different philosophy.