Identify intervals within a specified distance.
Arguments
- x
- y
ivl_df, or a path or URL to a bigWig (
.bw) or bigBed (.bb) file. When a file is supplied, only the windowed regions aroundxare read from it (local files andhttp(s)://URLs are both supported), avoiding the cost of loading the entire file.- genome
- ...
params for bed_slop and bed_intersect
Details
input tbls are grouped by chrom by default, and additional
groups can be added using dplyr::group_by(). For example,
grouping by strand will constrain analyses to the same strand. To
compare opposing strands across two tbls, strands on the y tbl can
first be inverted using flip_strands().
See also
https://bedtools.readthedocs.io/en/latest/content/tools/window.html
Other multiple set operations:
bed_closest(),
bed_coverage(),
bed_intersect(),
bed_map(),
bed_subtract()
Examples
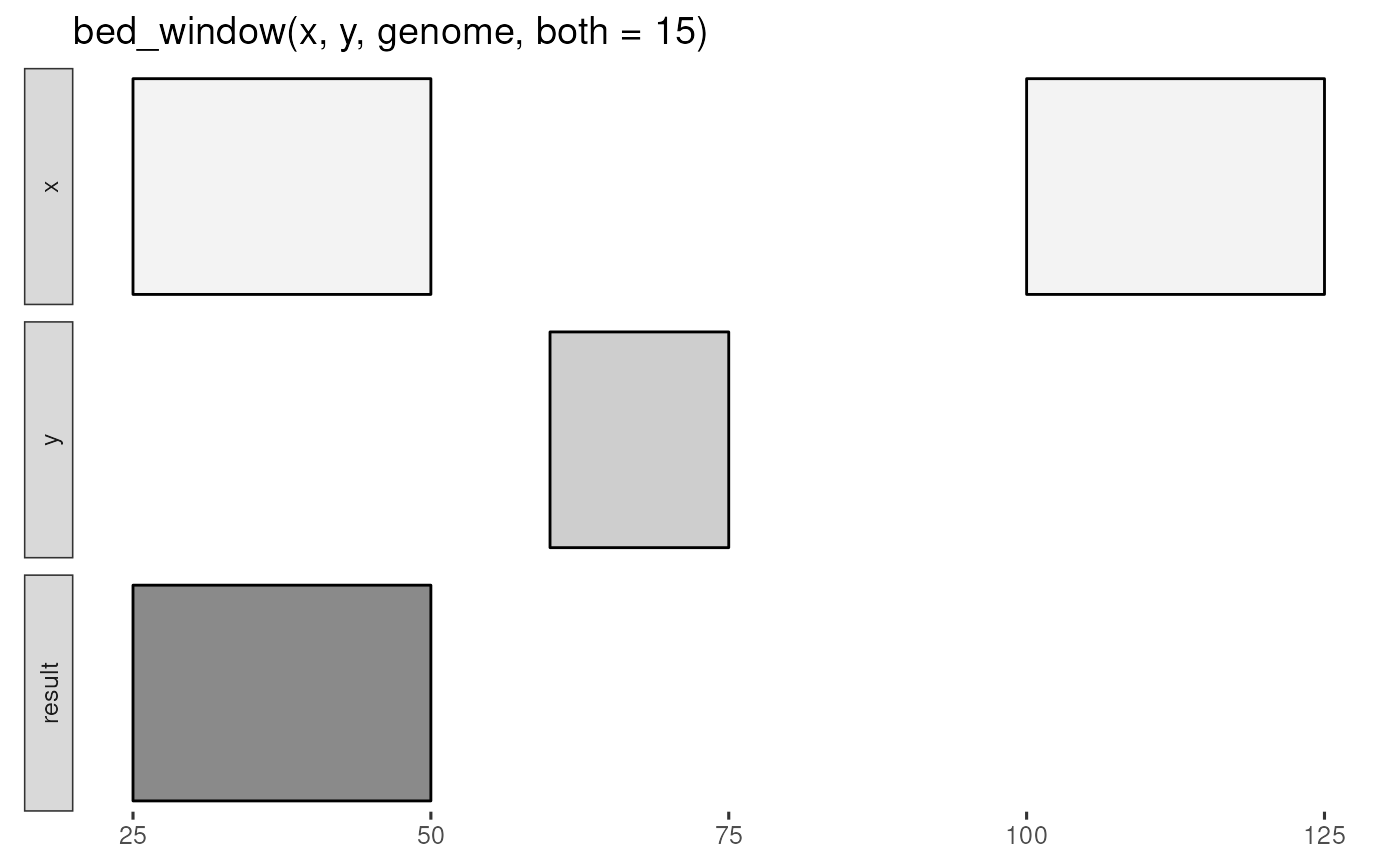
x <- tibble::tribble(
~chrom, ~start, ~end,
"chr1", 25, 50,
"chr1", 100, 125
)
y <- tibble::tribble(
~chrom, ~start, ~end,
"chr1", 60, 75
)
genome <- tibble::tribble(
~chrom, ~size,
"chr1", 125
)
bed_glyph(bed_window(x, y, genome, both = 15))
 x <- tibble::tribble(
~chrom, ~start, ~end,
"chr1", 10, 100,
"chr2", 200, 400,
"chr2", 300, 500,
"chr2", 800, 900
)
y <- tibble::tribble(
~chrom, ~start, ~end,
"chr1", 150, 400,
"chr2", 230, 430,
"chr2", 350, 430
)
genome <- tibble::tribble(
~chrom, ~size,
"chr1", 500,
"chr2", 1000
)
bed_window(x, y, genome, both = 100)
#> # A tibble: 4 × 7
#> chrom start.x end.x start.y end.y .source .overlap
#> <chr> <dbl> <dbl> <dbl> <dbl> <chr> <int>
#> 1 chr2 200 400 230 430 1 200
#> 2 chr2 200 400 350 430 1 80
#> 3 chr2 300 500 230 430 1 200
#> 4 chr2 300 500 350 430 1 80
# `y` can be a bigWig/bigBed file path or `http(s)://` URL; only the
# windowed regions around `x` are read from the file
xf <- tibble::tribble(
~chrom, ~start, ~end,
"chr1", 4840000, 4841000
)
gf <- read_genome(valr_example("hg19.chrom.sizes.gz"))
bed_window(xf, valr_example("test.bb"), gf, both = 10000)
#> # A tibble: 1 × 16
#> chrom start.x end.x start.y end.y name.y score.y strand.y thickStart.y
#> <chr> <dbl> <dbl> <dbl> <dbl> <chr> <int> <chr> <int>
#> 1 chr1 4840000 4841000 4797973 4836816 testgene 1 + 4797973
#> # ℹ 7 more variables: thickEnd.y <int>, reserved.y <int>, blockCount.y <int>,
#> # blockSizes.y <chr>, chromStarts.y <chr>, .source <chr>, .overlap <int>
# add a `.dist` column to the output
if (FALSE) { # \dontrun{
bed_window(x, y, genome, both = 200) |>
mutate(
.dist = case_when(
.overlap == 0 ~ abs(pmax(start.x, start.y) - pmin(end.x, end.y)),
.default = 0
)
)
} # }
x <- tibble::tribble(
~chrom, ~start, ~end,
"chr1", 10, 100,
"chr2", 200, 400,
"chr2", 300, 500,
"chr2", 800, 900
)
y <- tibble::tribble(
~chrom, ~start, ~end,
"chr1", 150, 400,
"chr2", 230, 430,
"chr2", 350, 430
)
genome <- tibble::tribble(
~chrom, ~size,
"chr1", 500,
"chr2", 1000
)
bed_window(x, y, genome, both = 100)
#> # A tibble: 4 × 7
#> chrom start.x end.x start.y end.y .source .overlap
#> <chr> <dbl> <dbl> <dbl> <dbl> <chr> <int>
#> 1 chr2 200 400 230 430 1 200
#> 2 chr2 200 400 350 430 1 80
#> 3 chr2 300 500 230 430 1 200
#> 4 chr2 300 500 350 430 1 80
# `y` can be a bigWig/bigBed file path or `http(s)://` URL; only the
# windowed regions around `x` are read from the file
xf <- tibble::tribble(
~chrom, ~start, ~end,
"chr1", 4840000, 4841000
)
gf <- read_genome(valr_example("hg19.chrom.sizes.gz"))
bed_window(xf, valr_example("test.bb"), gf, both = 10000)
#> # A tibble: 1 × 16
#> chrom start.x end.x start.y end.y name.y score.y strand.y thickStart.y
#> <chr> <dbl> <dbl> <dbl> <dbl> <chr> <int> <chr> <int>
#> 1 chr1 4840000 4841000 4797973 4836816 testgene 1 + 4797973
#> # ℹ 7 more variables: thickEnd.y <int>, reserved.y <int>, blockCount.y <int>,
#> # blockSizes.y <chr>, chromStarts.y <chr>, .source <chr>, .overlap <int>
# add a `.dist` column to the output
if (FALSE) { # \dontrun{
bed_window(x, y, genome, both = 200) |>
mutate(
.dist = case_when(
.overlap == 0 ~ abs(pmax(start.x, start.y) - pmin(end.x, end.y)),
.default = 0
)
)
} # }