Plot the usage of different V(D)J segments for each cell cluster. The usage of two V(D)J segments can also be plotted for a single chain.
Usage
plot_gene_usage(
input,
data_cols,
cluster_col = NULL,
group_col = NULL,
method = NULL,
units = "percent",
genes = 20,
return_list = FALSE,
plot_colors = NULL,
plot_lvls = NULL,
trans = "identity",
rotate_labels = FALSE,
panel_nrow = NULL,
show_points = TRUE,
show_zeros = TRUE,
n_label = NULL,
p_label = c(value = 0.05),
p_method = NULL,
p_file = NULL,
label_params = list(),
...,
chain = NULL,
chain_col = global$chain_col,
sep = global$sep
)Arguments
- input
Object containing V(D)J data. If a data.frame is provided, the cell barcodes should be stored as row names.
- data_cols
meta.data column containing genes for each clonotype, provide a vector with two column names to plot paired usage of genes
- cluster_col
meta.data column containing cell clusters to use for calculating gene usage
- group_col
meta.data column to use for grouping cluster IDs present in cluster_col. This is useful when there are multiple replicates or patients for each treatment condition.
- method
Method to use for plotting, possible values are:
'bar', create a bargraph, this is the default when a single column is passed to the data_cols argument
'boxplot', create boxplots, this can only be used when group_col is provided
'heatmap', create a heatmap, this is the default when two columns are passed to the data_cols argument
'circos', create a circos plot, this requires two columns to be provided to the data_cols argument
- units
Units to plot on the y-axis, either 'frequency' or 'percent'
- genes
An integer specifying the number of genes to plot, or a vector giving the names of genes to include.
- return_list
Should a list of plots be returned, if FALSE plots will be combined and arranged into panels
- plot_colors
Character vector containing colors to use for plot. If a bar graph is created this will specify how to color cell clusters. For a heatmap, these colors will be used to generate the color gradient.
- plot_lvls
Levels to use for ordering clusters
- trans
Transformation to use when plotting segment usage, e.g. 'log10'. By default values are not transformed, refer to
ggplot2::continuous_scale()for more options.- rotate_labels
Should labels on circos plot be rotated to reduce overlapping text
- panel_nrow
The number of rows to use for arranging plots when return_list is FALSE
- show_points
If
TRUEdata points will be shown on boxplots, the point size can be adjusted using thepoint.sizeparameter- show_zeros
If
TRUEcell labels that are missing from a cluster will still be shown on the plot- n_label
Location on plot where n label should be added, this is only applicable when
methodis 'bar' and can be any combination of the following:'corner', display the total number of cells plotted in the top right corner, the position of the label can be modified by passing
xandyspecifications with thelabel_paramsargument'legend', display the number of cells plotted for each group shown in the plot legend
'none', do not display the number of cells plotted
- p_label
Specification indicating how p-values should be labeled on plot, this can one of the following:
'none', do not display p-values
'all', show p-values for all groups
A named vector providing p-value cutoffs and labels to display, e.g.
c('*' = 0.05, '**' = 0.01, '***' = 0.001). The keyword 'value' can be used to display the p-value for those less than a certain cutoff, e.g.c(value = 0.05, ns = Inf)will show significant p-values, all others will be labeled 'ns'.
- p_method
Method to use for calculating p-values. By default when comparing two groups a t-test will be performed, when comparing more than two groups the Kruskal-Wallis test will be used. p-values are adjusted for multiple testing using Bonferroni correction. Possible methods include:
't', two sample t-test performed with
stats::t.test()'wilcox', Wilcoxon rank sum test performed with
stats::wilcox.test()'kruskal', Kruskal-Wallis test performed with
stats::kruskal.test()
- p_file
File path to save table containing p-values for each comparison.
- label_params
Named list providing additional parameters to modify n label aesthetics, e.g. list(size = 4, color = "red")
- ...
Additional arguments to pass to plotting function,
ggplot2::geom_col()for bargraph,ggplot2::geom_tile()for heatmap,circlize::chordDiagram()for circos plot- chain
Chain to use for calculating gene usage, set to NULL to include all chains
- chain_col
meta.data column containing chains for each cell
- sep
Separator used for storing per-chain V(D)J data for each cell
Examples
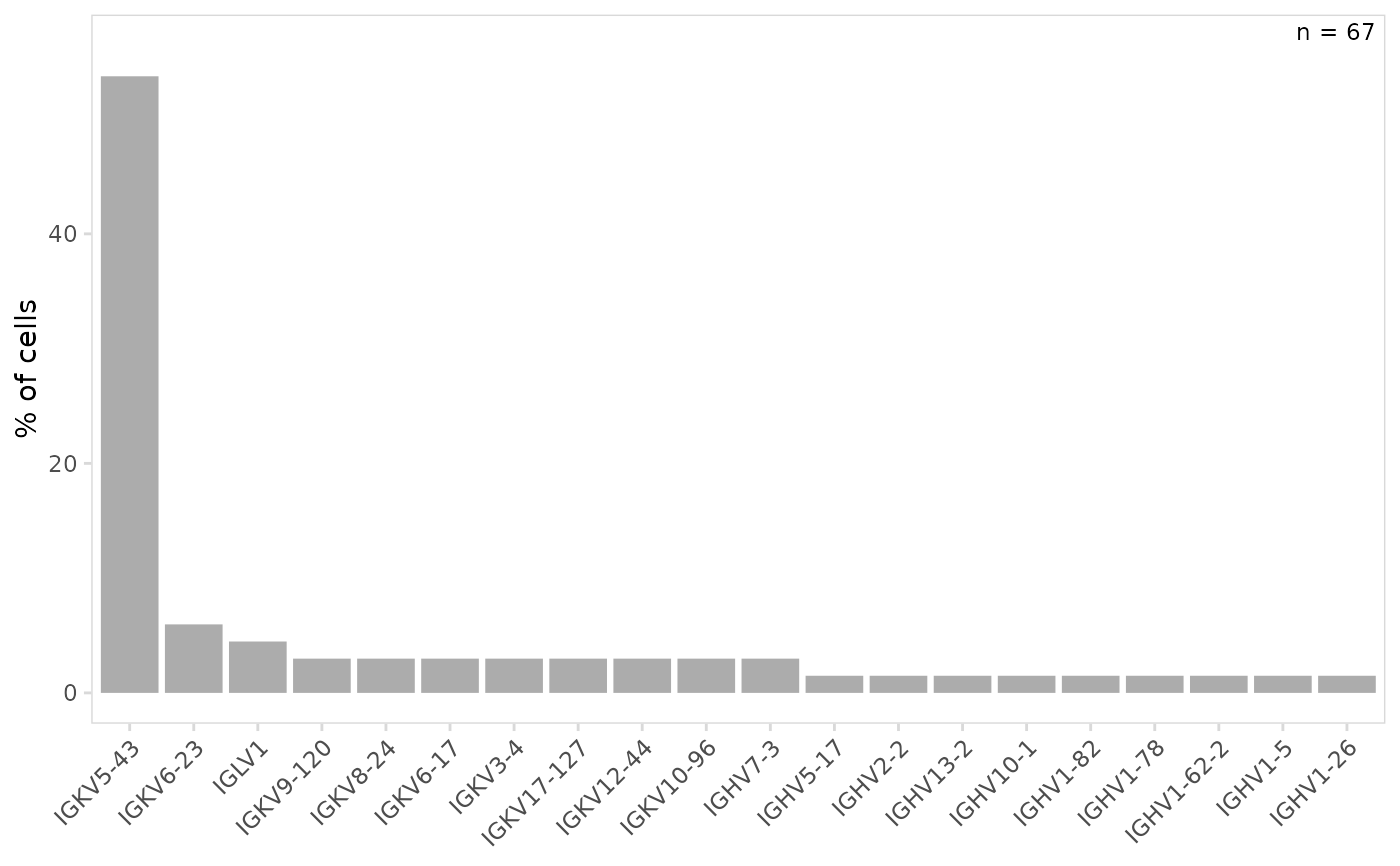
# Plot V(D)J segment usage for all cells
plot_gene_usage(
vdj_sce,
data_cols = "v_gene"
)
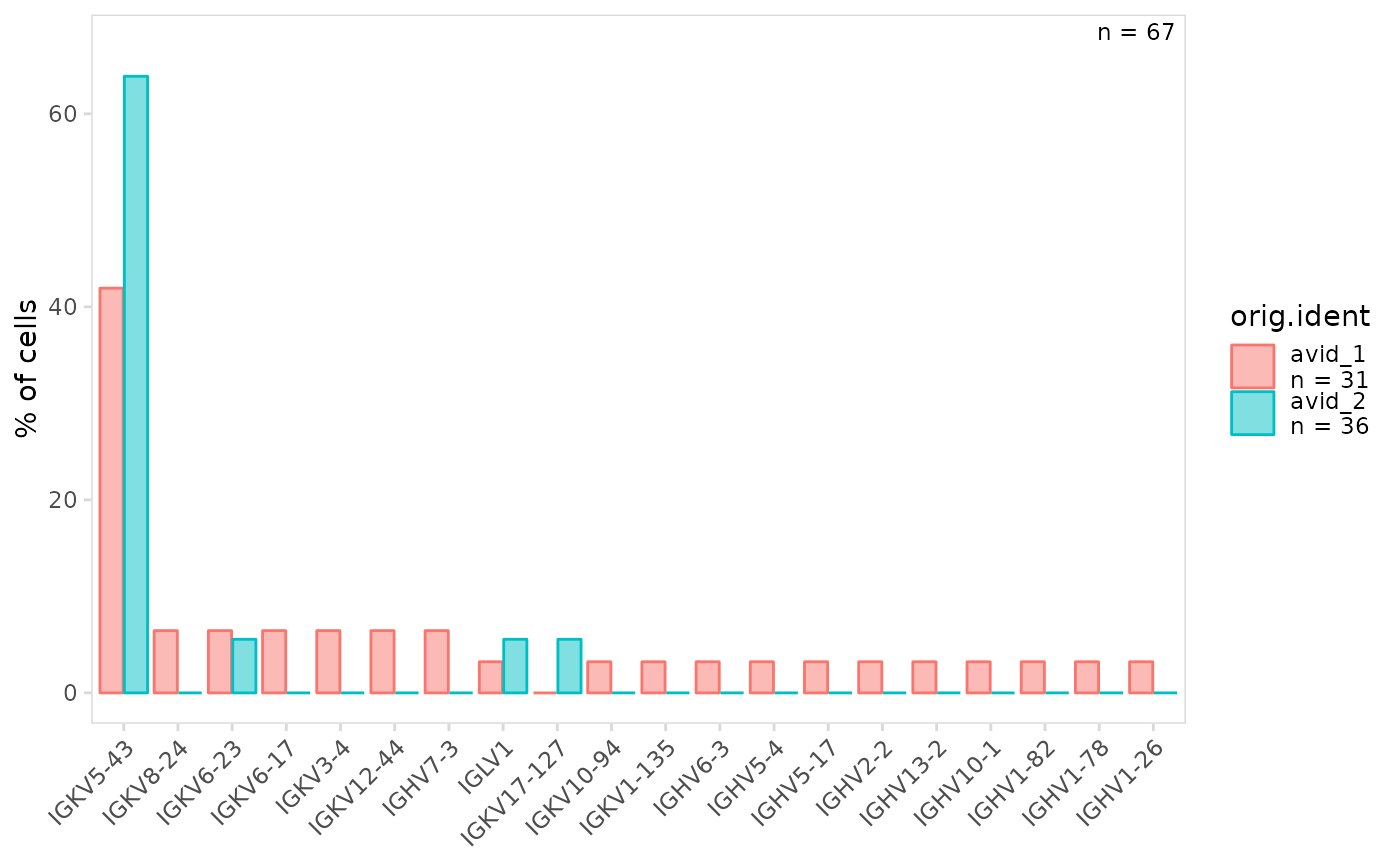
 # Plot gene usage separately for cell clusters
plot_gene_usage(
vdj_sce,
data_cols = "v_gene",
cluster_col = "orig.ident"
)
# Plot gene usage separately for cell clusters
plot_gene_usage(
vdj_sce,
data_cols = "v_gene",
cluster_col = "orig.ident"
)
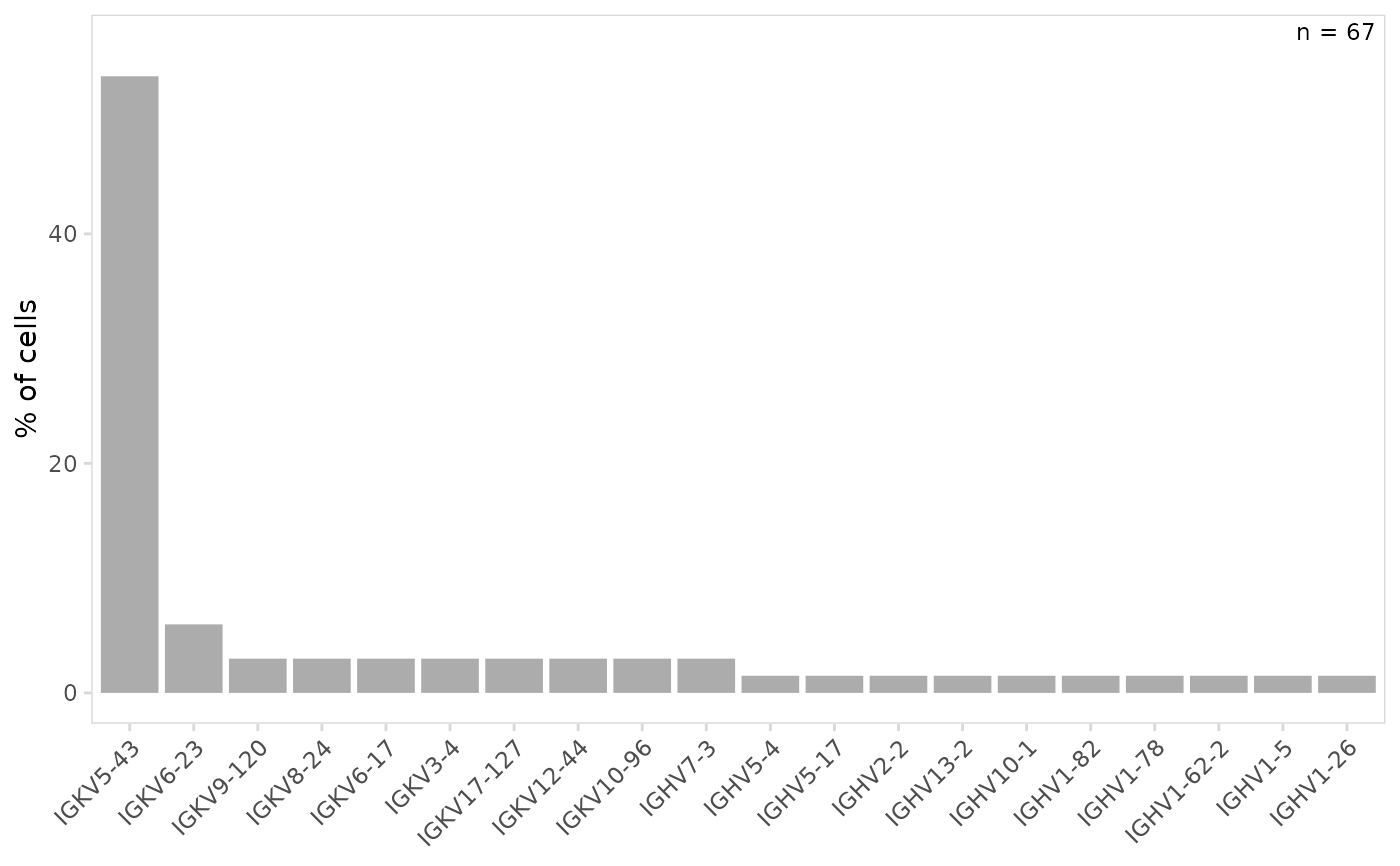
 # Plot gene usage for a specific chain
plot_gene_usage(
vdj_sce,
data_cols = "v_gene",
chain = c("IGH", "IGK")
)
# Plot gene usage for a specific chain
plot_gene_usage(
vdj_sce,
data_cols = "v_gene",
chain = c("IGH", "IGK")
)
 # Plot paired usage of V(D)J segments
plot_gene_usage(
vdj_sce,
data_cols = c("v_gene", "j_gene"),
type = "circos"
)
#> Warning: Ignoring unknown parameters: `type`
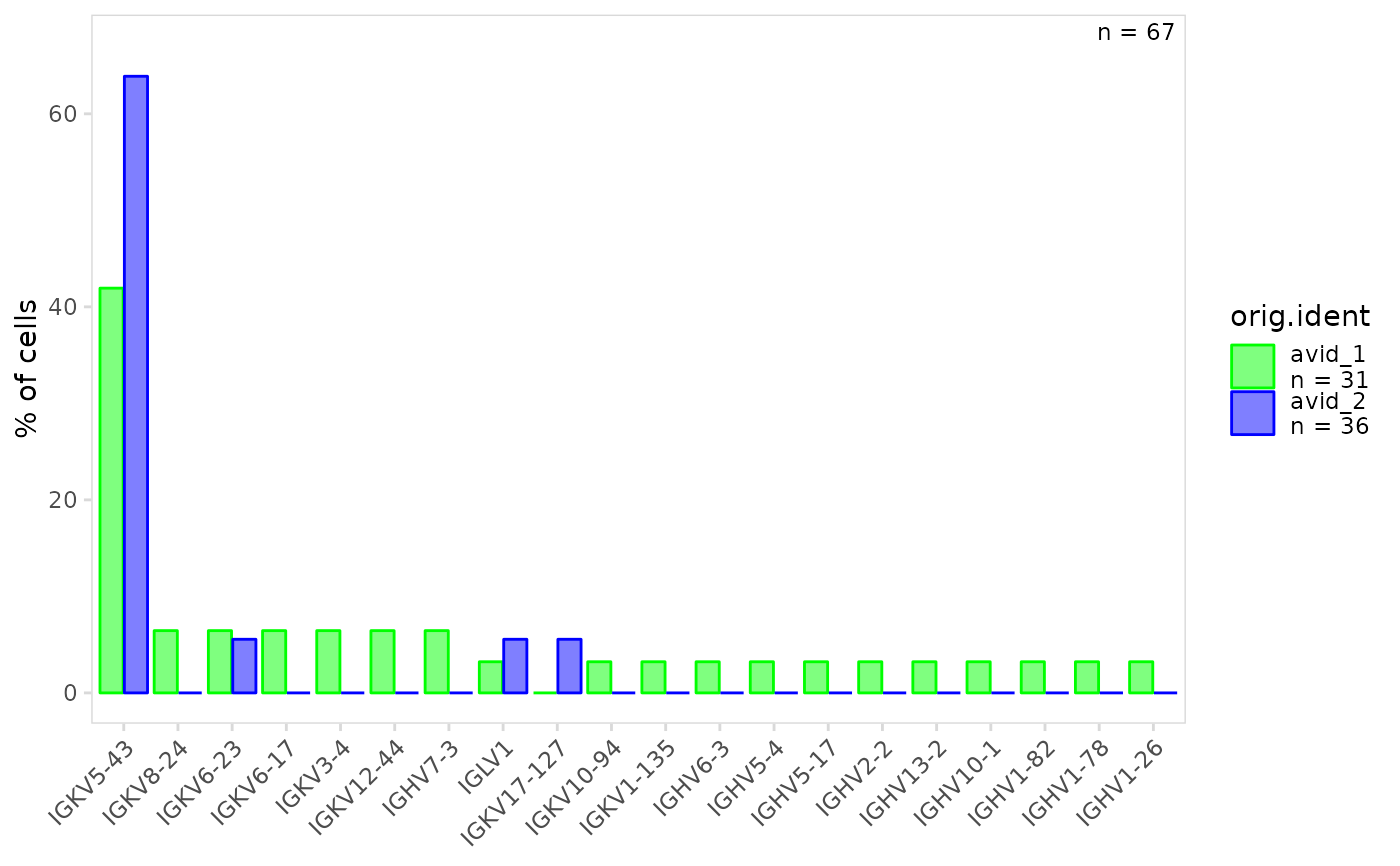
# Specify colors to use for each cell cluster
plot_gene_usage(
vdj_sce,
data_cols = "v_gene",
cluster_col = "orig.ident",
plot_colors = c(avid_2 = "blue", avid_1 = "green")
)
# Plot paired usage of V(D)J segments
plot_gene_usage(
vdj_sce,
data_cols = c("v_gene", "j_gene"),
type = "circos"
)
#> Warning: Ignoring unknown parameters: `type`
# Specify colors to use for each cell cluster
plot_gene_usage(
vdj_sce,
data_cols = "v_gene",
cluster_col = "orig.ident",
plot_colors = c(avid_2 = "blue", avid_1 = "green")
)
 # Specify order to use for plotting cell clusters
plot_gene_usage(
vdj_sce,
data_cols = "v_gene",
cluster_col = "orig.ident",
plot_lvls = c("avid_2", "avid_1")
)
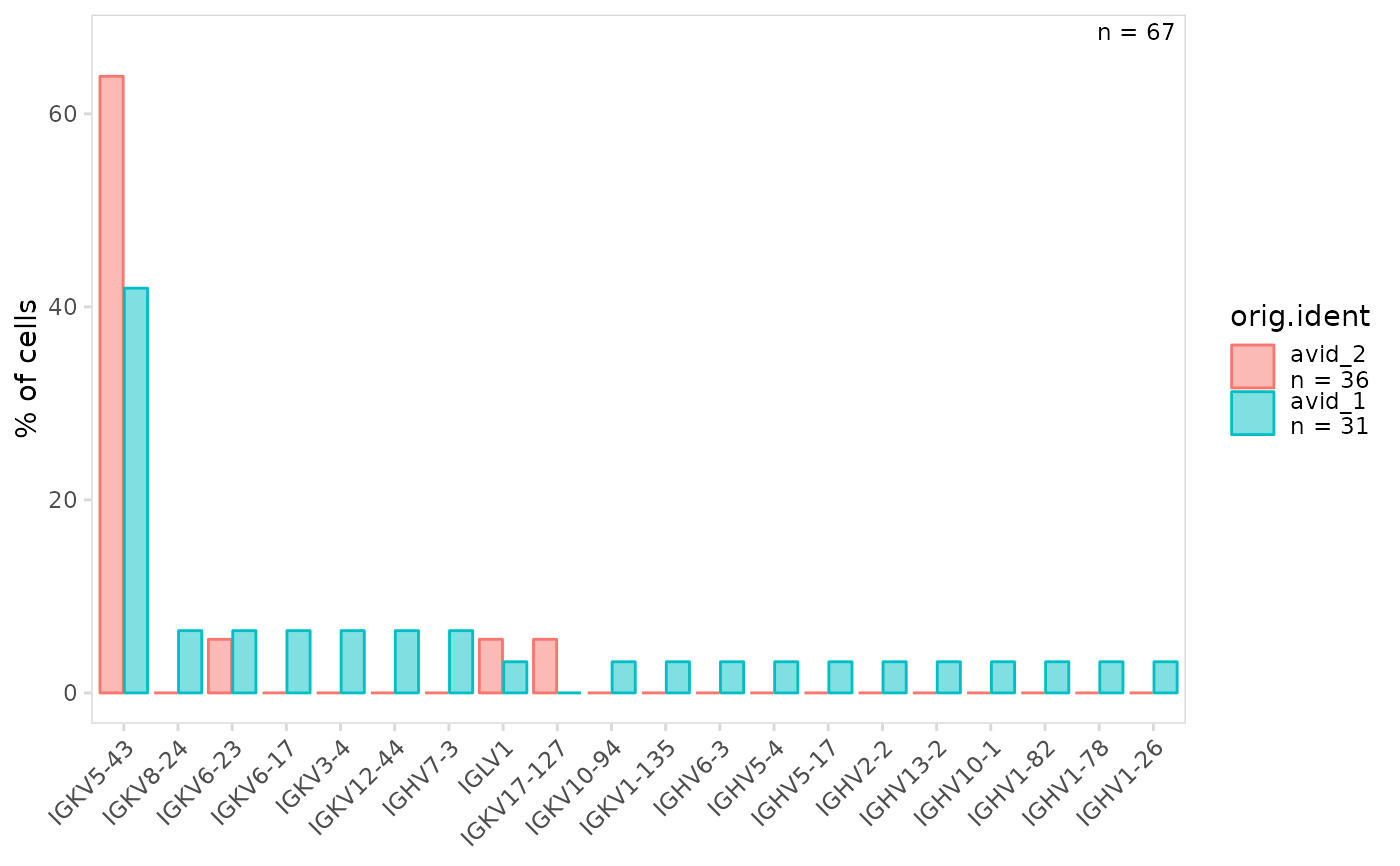
# Specify order to use for plotting cell clusters
plot_gene_usage(
vdj_sce,
data_cols = "v_gene",
cluster_col = "orig.ident",
plot_lvls = c("avid_2", "avid_1")
)
 # Specify certain V(D)J genes to include in plot
plot_gene_usage(
vdj_sce,
data_cols = "v_gene",
vdj_genes = c("IGKV5-43", "IGLV1", "IGHV1-64")
)
#> Warning: Ignoring unknown parameters: `vdj_genes`
# Specify certain V(D)J genes to include in plot
plot_gene_usage(
vdj_sce,
data_cols = "v_gene",
vdj_genes = c("IGKV5-43", "IGLV1", "IGHV1-64")
)
#> Warning: Ignoring unknown parameters: `vdj_genes`
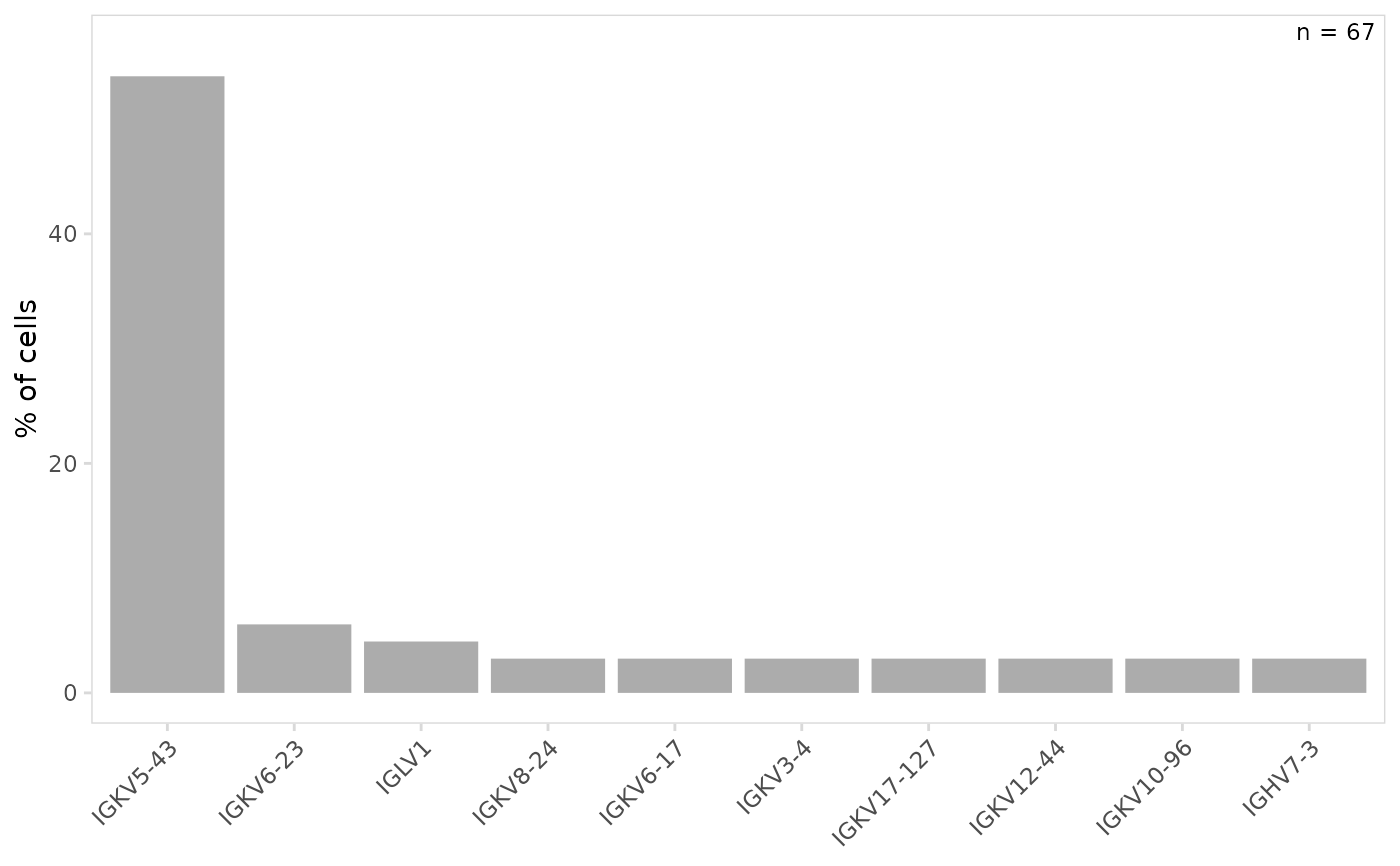
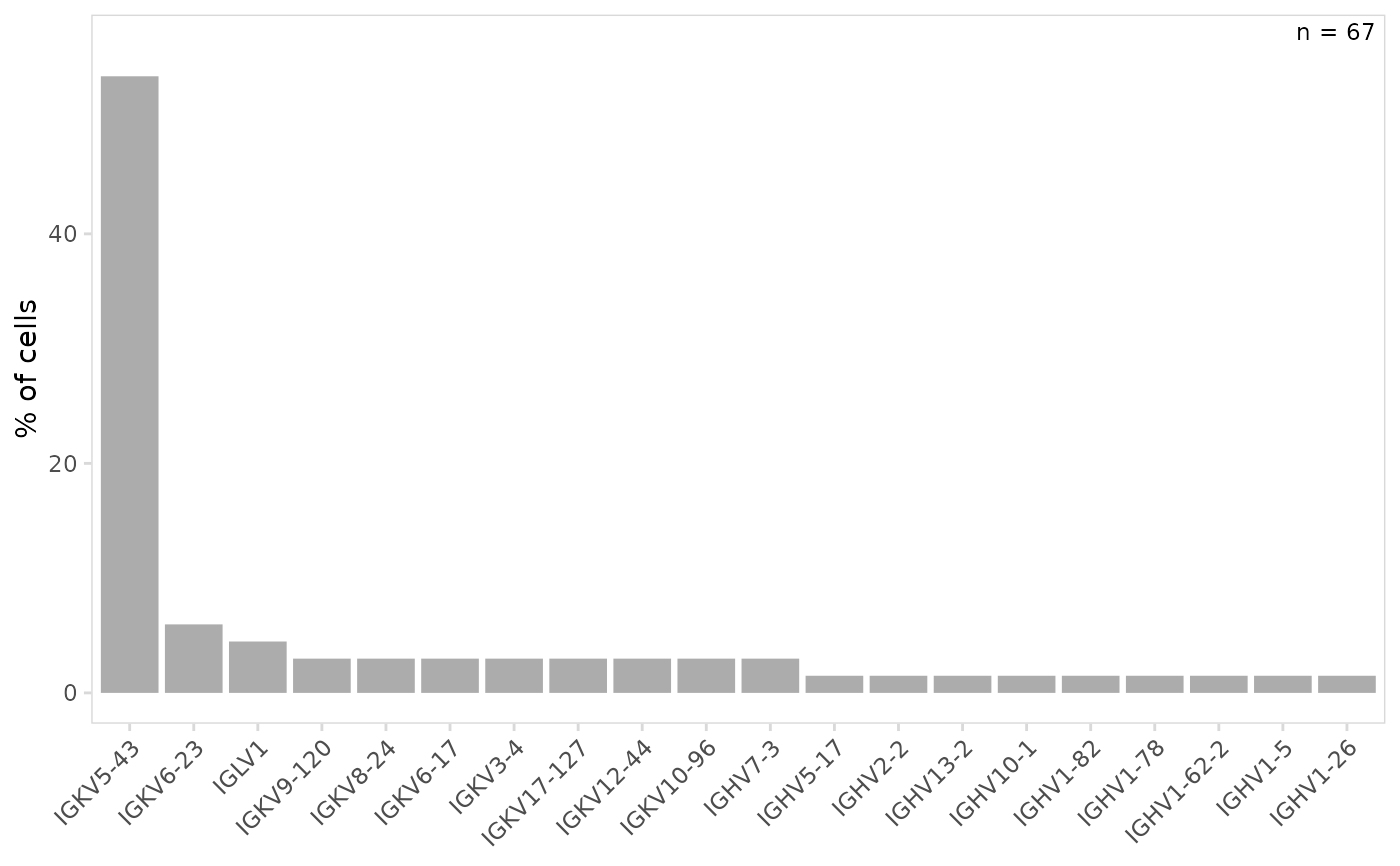 # Specify the number of top V(D)J genes to include in plot
plot_gene_usage(
vdj_sce,
data_cols = "v_gene",
genes = 10
)
# Specify the number of top V(D)J genes to include in plot
plot_gene_usage(
vdj_sce,
data_cols = "v_gene",
genes = 10
)