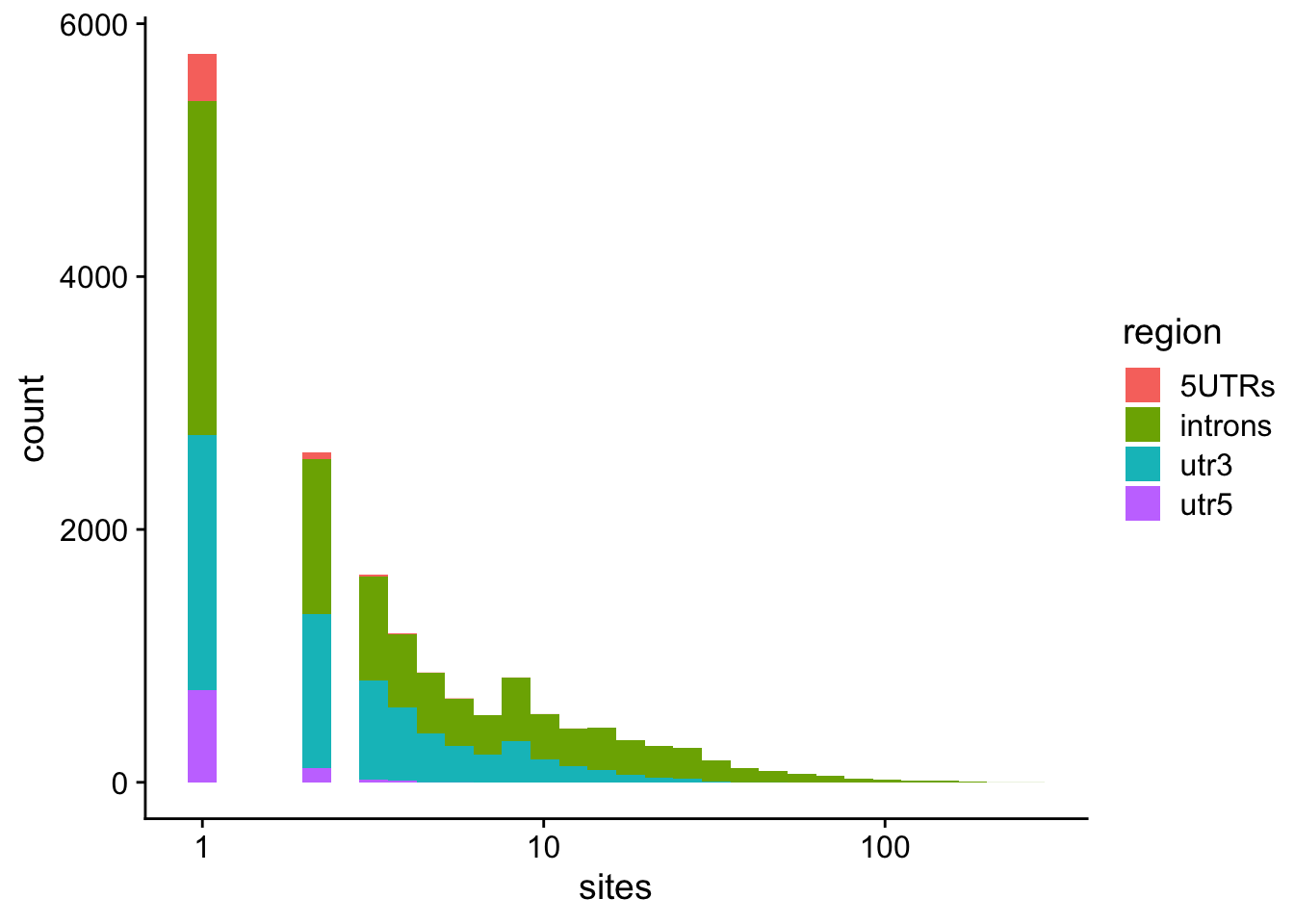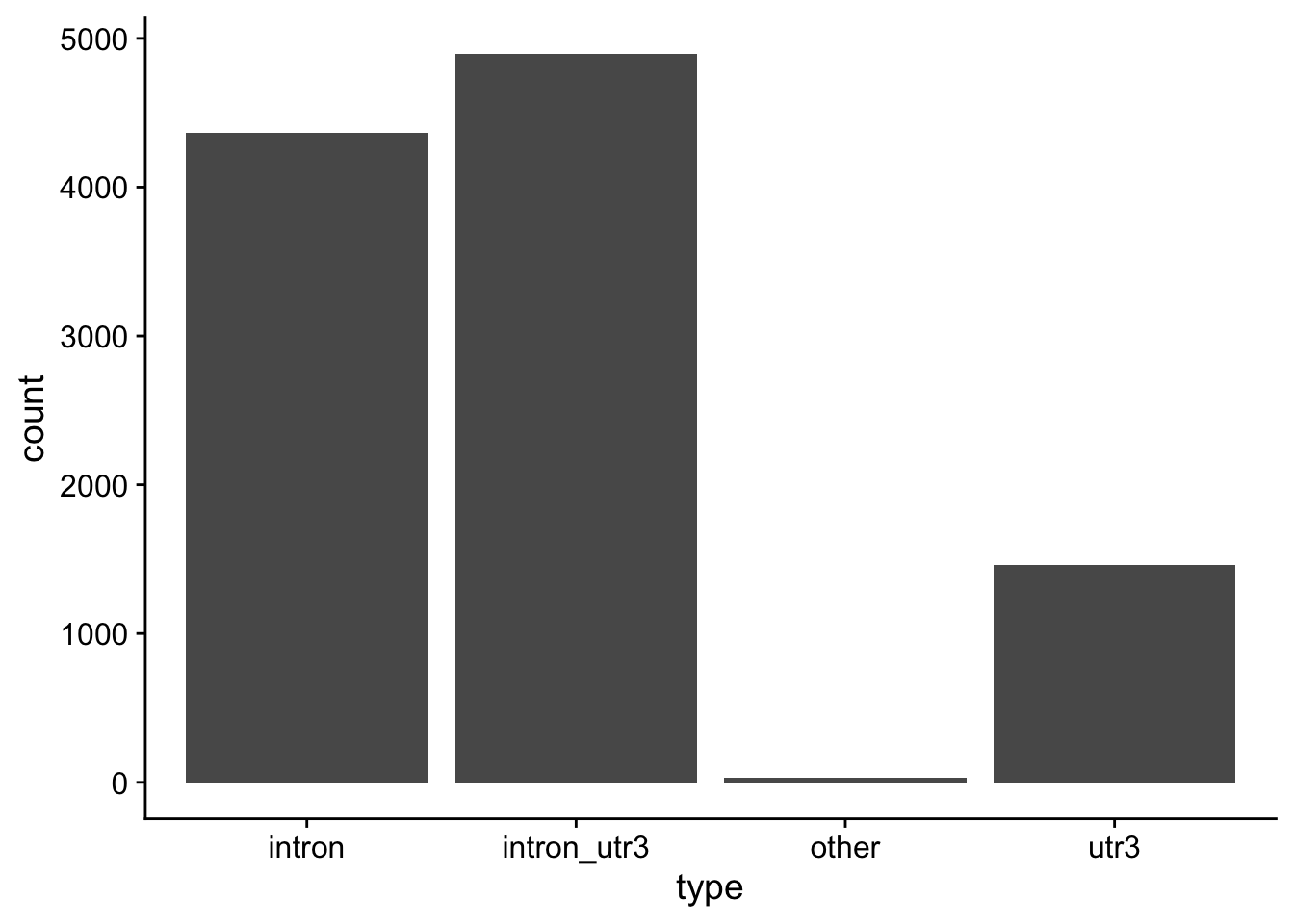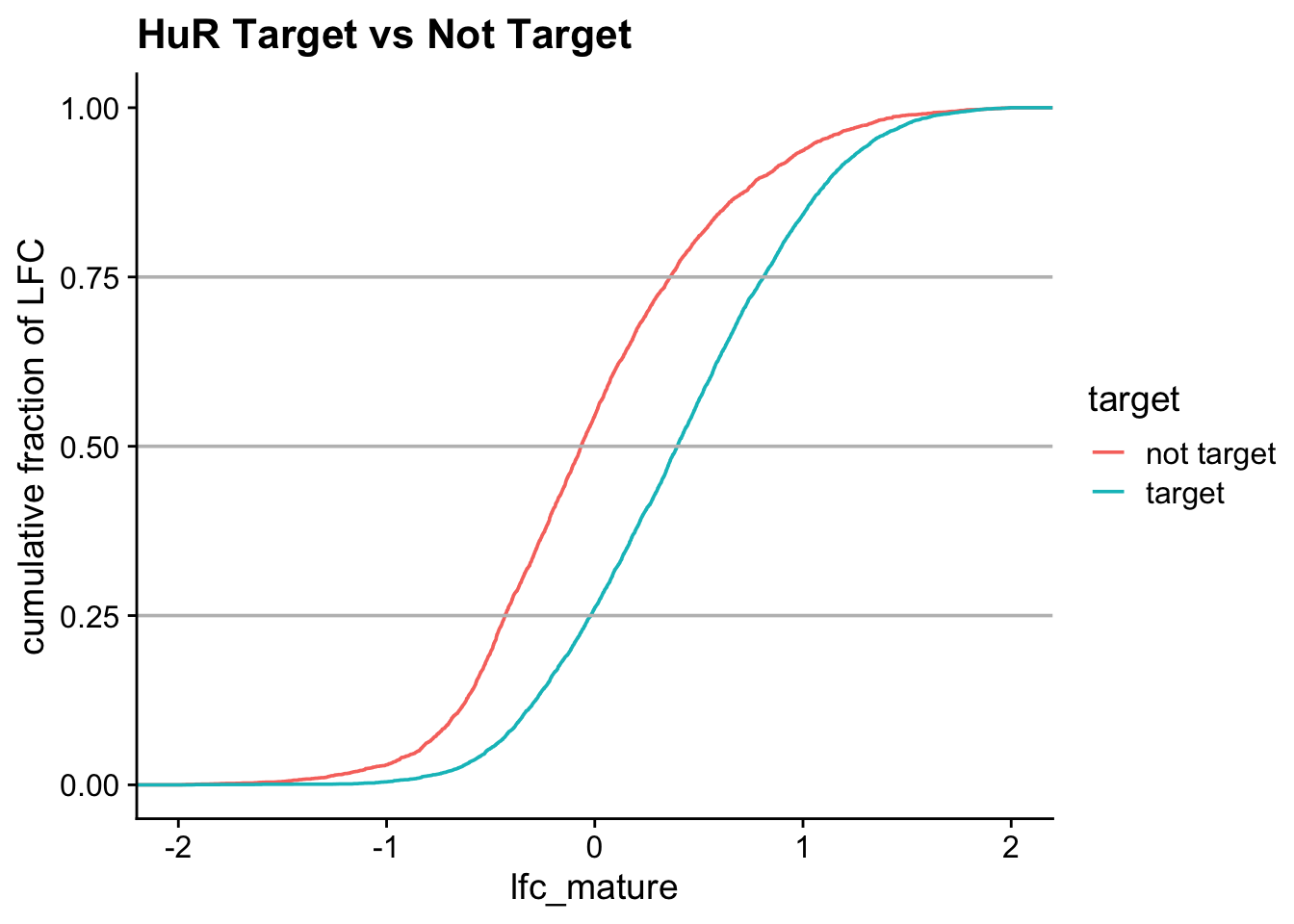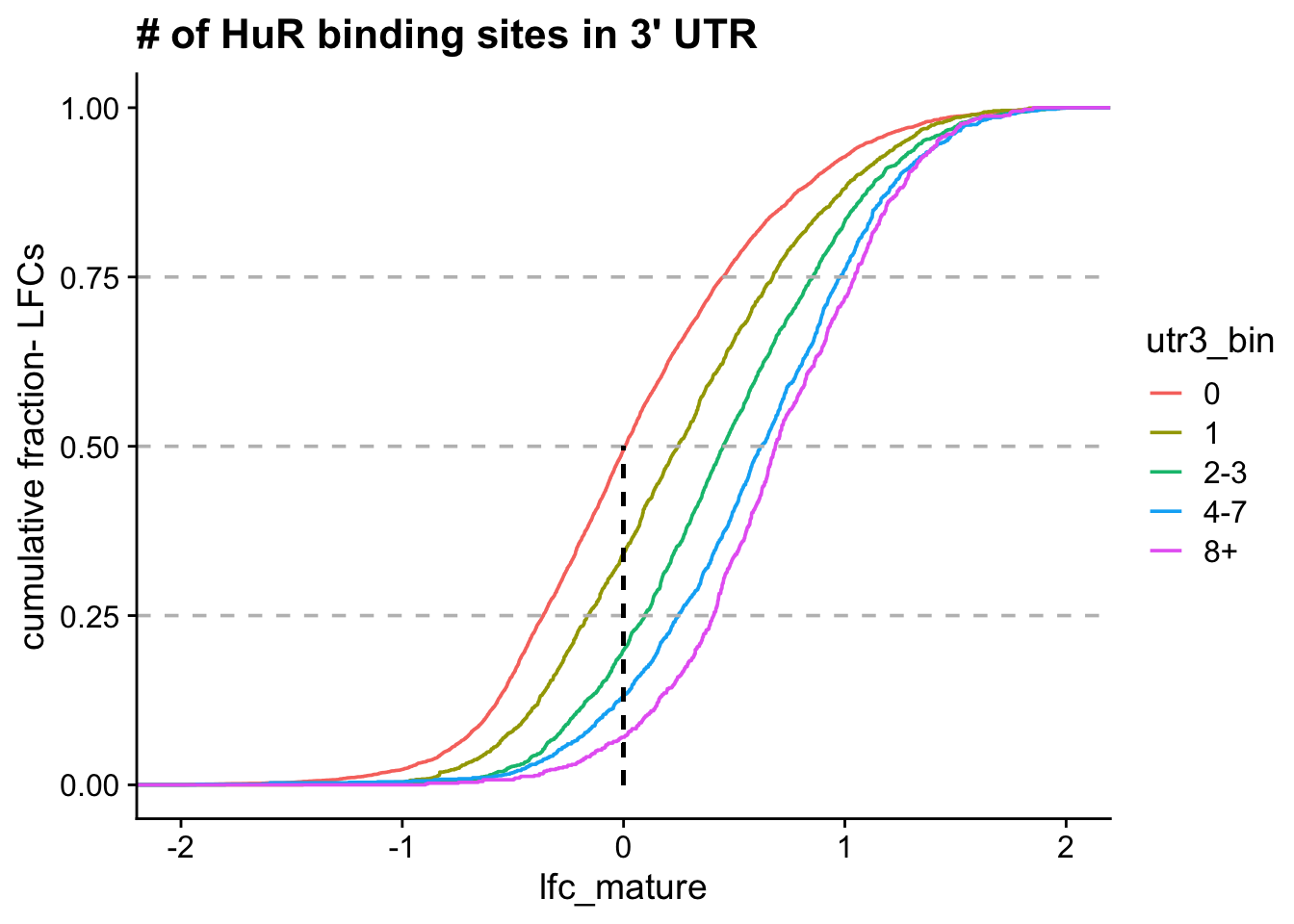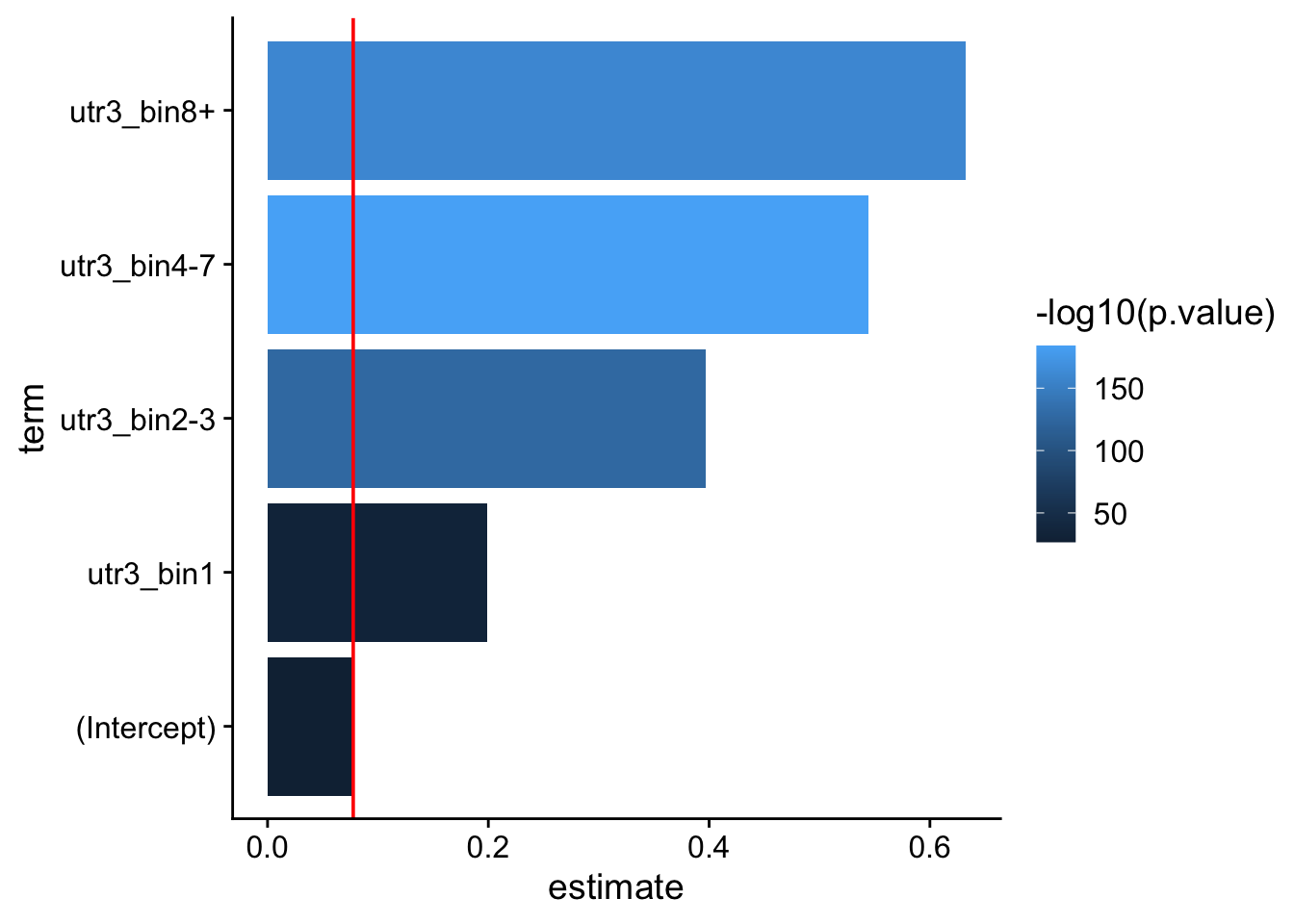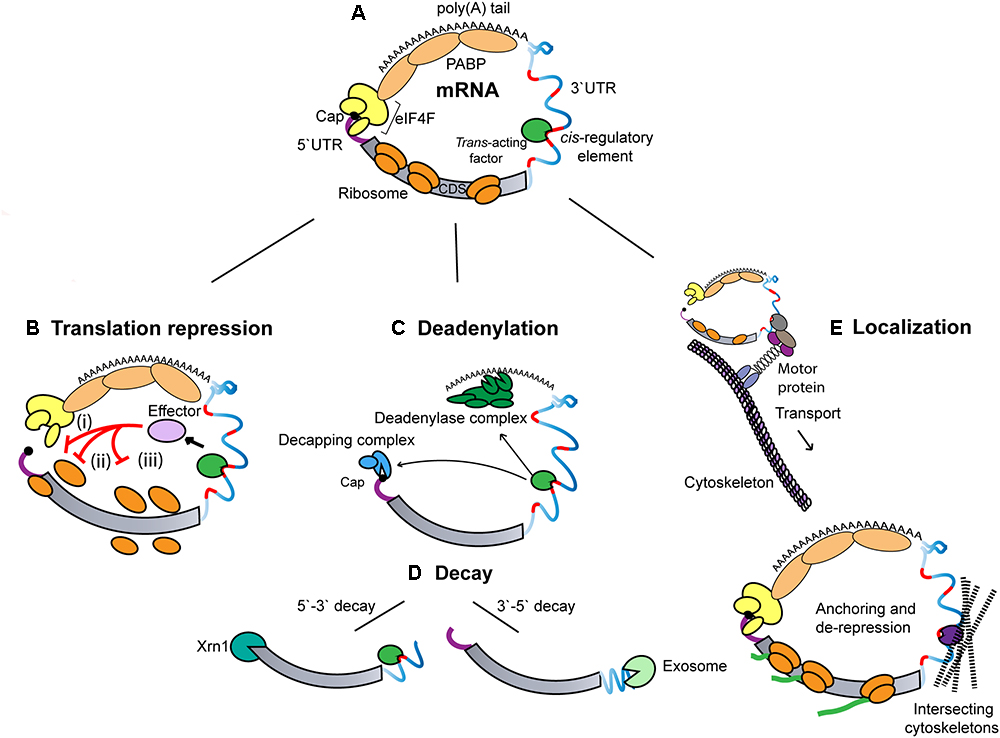
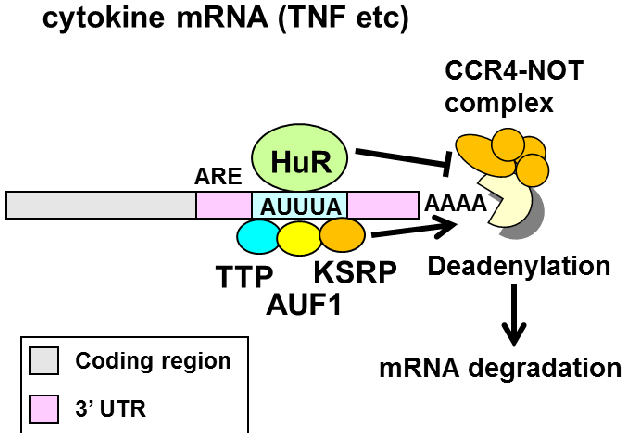
 Model predictions:
Model predictions:possible_annotations <- builtin_annotations()
# grep to keep those containing "hg19"
hg19_annots <- grep("hg19_genes", possible_annotations, value = T)
# WHY DID WE PICK hg19?
# let's keep 5' utr, cds, intron, 3' utr and intergenic
my_hg19_annots <- hg19_annots[c(3, 4, 7, 10, 11)]
# build the annotation database
annotations <- build_annotations(genome = "hg19", annotations = my_hg19_annots)'select()' returned 1:1 mapping between keys and
columnsBuilding promoters...Building 1to5kb upstream of TSS...Building intergenic...Building cds...Building 5UTRs...Building 3UTRs...Building exons...Building introns...GRanges object with 3067854 ranges and 5 metadata columns:
seqnames ranges strand |
<Rle> <IRanges> <Rle> |
[1] chr1 65565-65573 + |
[2] chr1 69037-70008 + |
[3] chr1 367659-368597 + |
[4] chr1 859812-860328 + |
[5] chr1 861302-861393 + |
... ... ... ... .
[3067850] chr21_gl383580_alt 1-74652 * |
[3067851] chr21_gl383581_alt 1-116690 * |
[3067852] chr22_gl383582_alt 1-162811 * |
[3067853] chr22_gl383583_alt 1-96924 * |
[3067854] chr22_kb663609_alt 1-74013 * |
id tx_id
<character> <character>
[1] CDS:1 ENST00000641515.2_6
[2] CDS:2 ENST00000641515.2_6
[3] CDS:3 ENST00000426406.1
[4] CDS:4 ENST00000616016.5_7
[5] CDS:5 ENST00000616016.5_7
... ... ...
[3067850] intergenic:22783 <NA>
[3067851] intergenic:22784 <NA>
[3067852] intergenic:22785 <NA>
[3067853] intergenic:22786 <NA>
[3067854] intergenic:22787 <NA>
gene_id symbol type
<character> <character> <character>
[1] 79501 OR4F5 hg19_genes_cds
[2] 79501 OR4F5 hg19_genes_cds
[3] 729759 OR4F29 hg19_genes_cds
[4] 148398 SAMD11 hg19_genes_cds
[5] 148398 SAMD11 hg19_genes_cds
... ... ... ...
[3067850] <NA> <NA> hg19_genes_intergenic
[3067851] <NA> <NA> hg19_genes_intergenic
[3067852] <NA> <NA> hg19_genes_intergenic
[3067853] <NA> <NA> hg19_genes_intergenic
[3067854] <NA> <NA> hg19_genes_intergenic
-------
seqinfo: 298 sequences (2 circular) from hg19 genome