Mapping chromatin structure and transactions
2026-03-27
Chromatin organization regulates genome compaction and gene expression
DNA accessibility drives all biochemical transactions on the genome
- Transcription Initiation
- Transcription Elongation
- DNA Repair
- Initiation of DNA Replication
- Recombination
- Viral Integration

Mapping & measurement are first steps toward understanding.
Before genome-wide DNA accessibility measurements, we knew about chromatin transactions at only a handful of loci.
This was a classic “keys under the lamppost” situation, leading to general models of chromatin-based gene regulation.

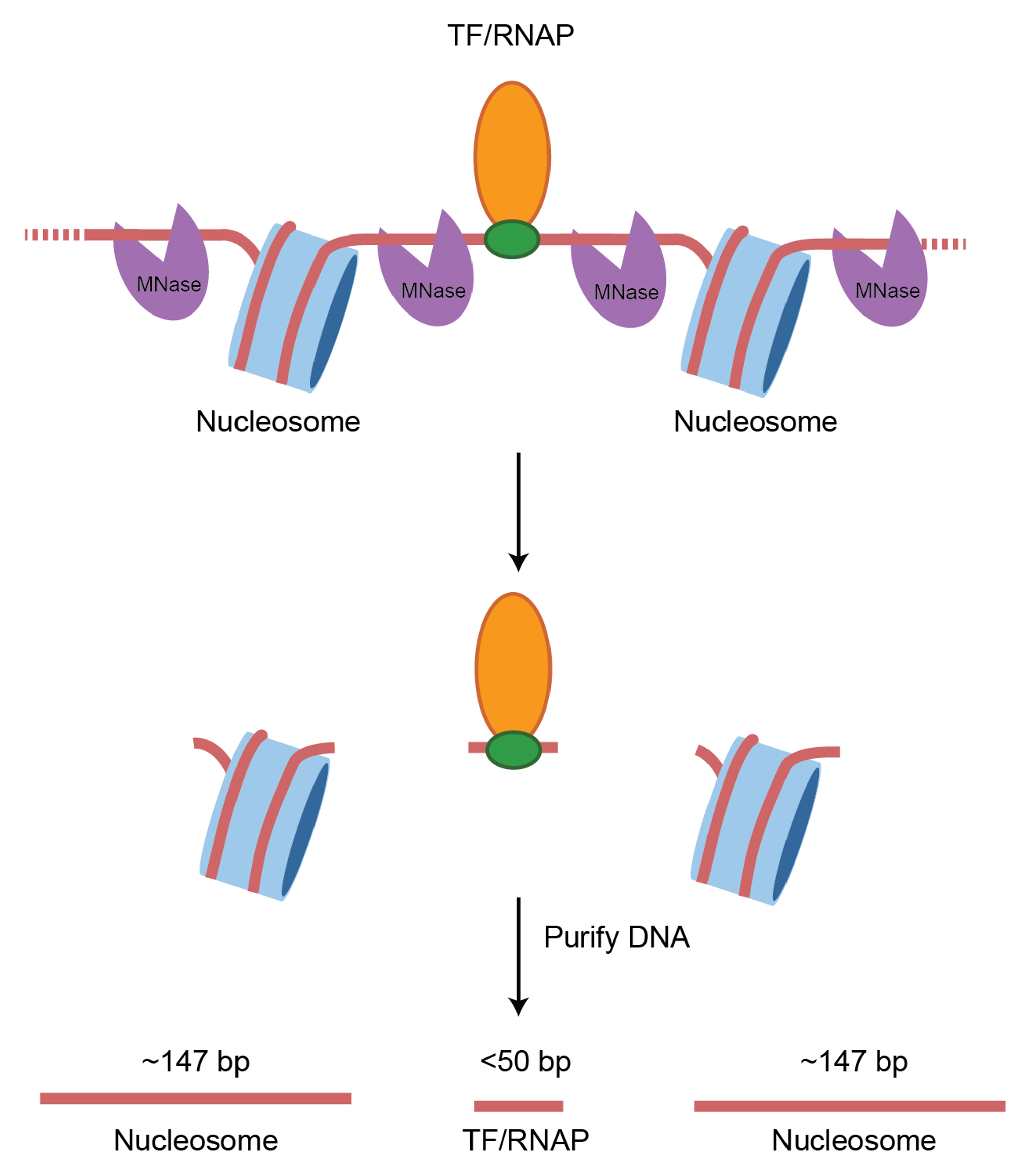
Using micrococcal nuclease (MNase) to map chromatin
- Micrococcal nuclease is an endo/exonuclease from Staphylococcus aureus
- Efficient, multiple turnover enzyme that digests accessible DNA (& RNA)
- Dependent on calcium ions
Using ATAC to map chromatin
- Tn5 transposase catalyzes “cut-and-paste” insertion of DNA into a target
- In ATAC (“Assay for Transposase-Accessible Chromatin”), the transposase enzymes are loaded with DNA sequencing adaptors (blue and red in the image), so the products of transposition are ready to PCR.
- Single turnover enzyme that acts on accessible DNA.
- Requires about ~60 bp of accessible for transposition.
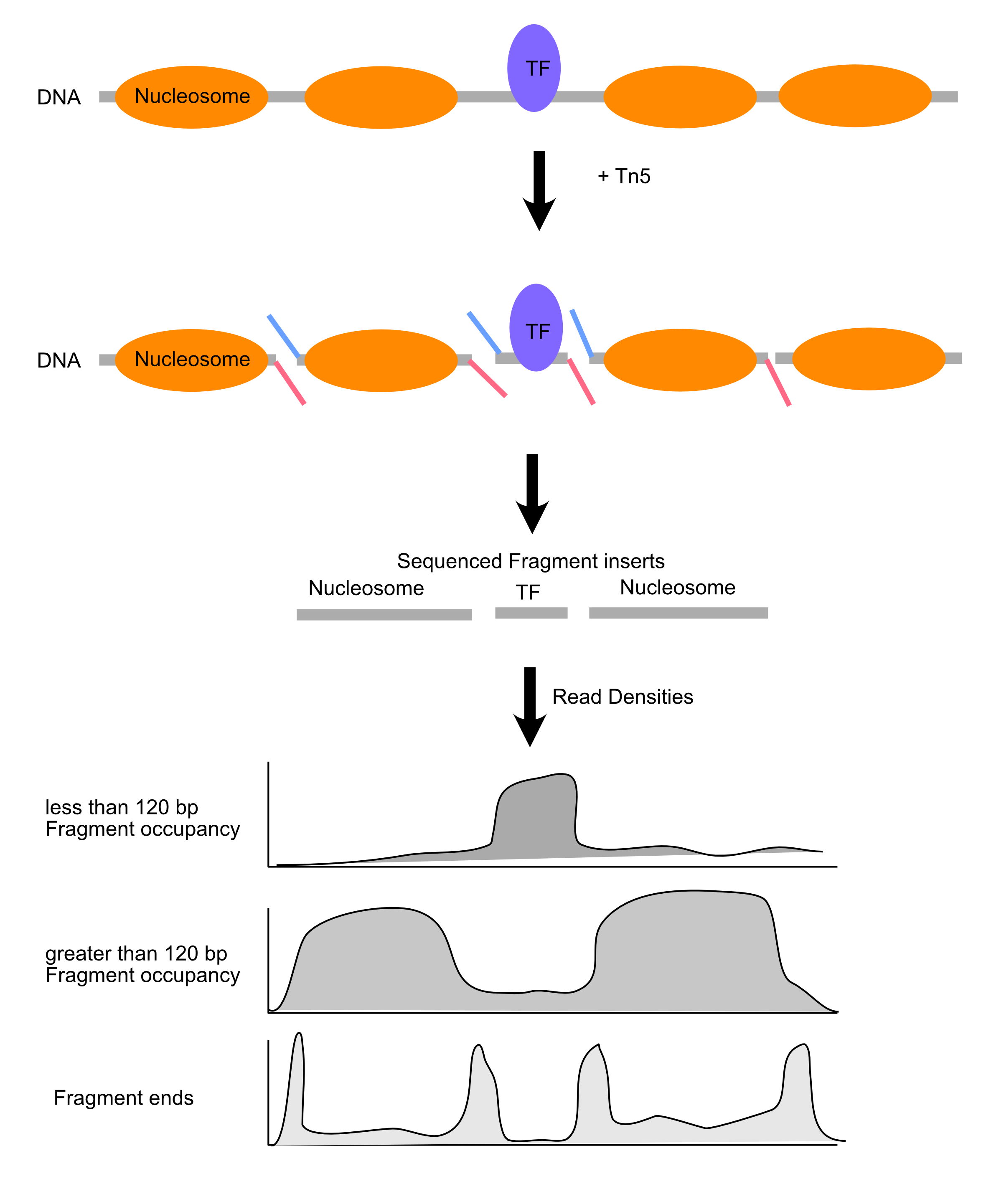


Experimental workflow

Interval analysis
The primary tool in the genome interval analysis is BEDtools – it’s the Swiss-army knife of internal analysis.
We will use an R package called valr that provides the same tools, but you don’t need to leave RStudio. valr provides the same tools for reading and manipulating genome intervals.
bed_intersect() is a fundamental operation. It identifies intervals from two tibbles that intersect and reports their overlaps.
Let’s take a look at that it does.
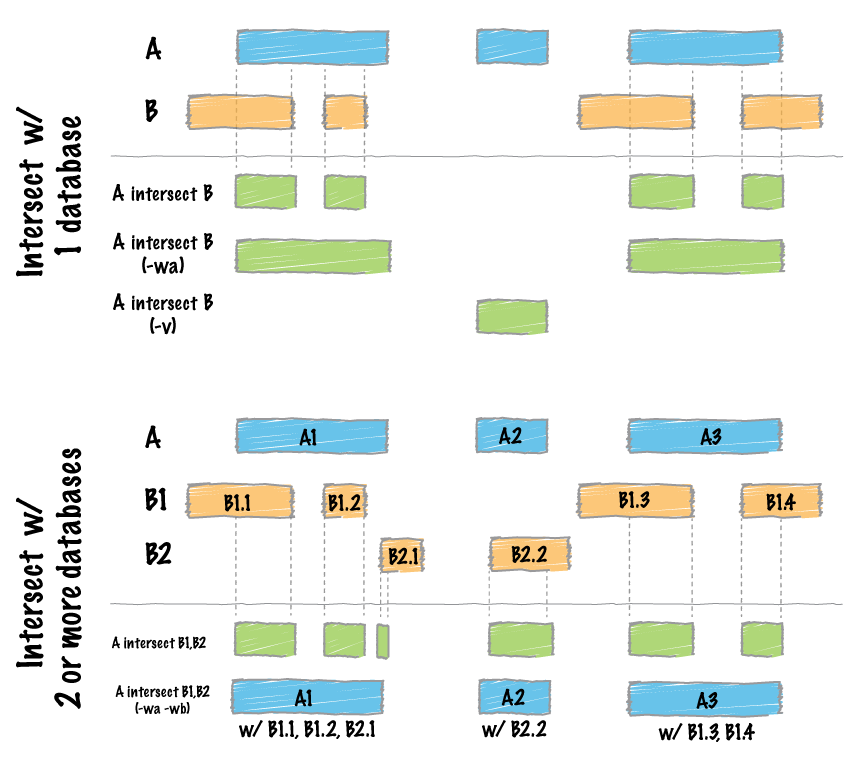
Visual representation of the intersecting intervals.

bed_map() example
bed_map() does two things in order:
- It identifies intersecting intervals between
xandy - Calculates summary statistics based on the intersection
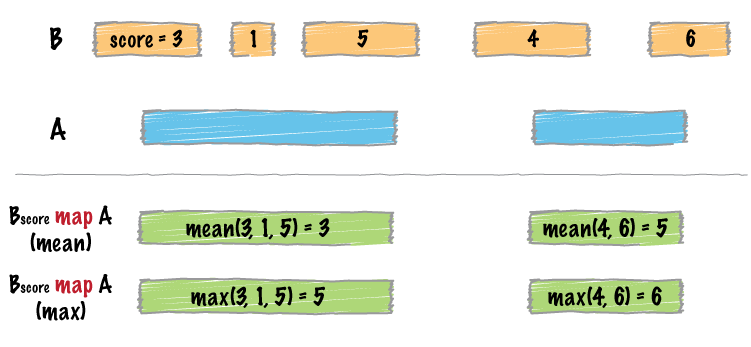
A typical use is to count up signals (e.g., coverage from an MNase-seq experiment) over specific regions (e.g., promoter regions).
Visual representation of bed_map()
