Problem Set Stats Bootcamp - class 14
Hypothesis Testing
biochem <- read_tsv(
"http://mtweb.cs.ucl.ac.uk/HSMICE/PHENOTYPES/Biochemistry.txt",
show_col_types = FALSE
) |>
janitor::clean_names()
# simplify names a bit more
colnames(biochem) <- gsub(
pattern = "biochem_",
replacement = "",
colnames(biochem)
)
# we are going to simplify this a bit and only keep some columns
keep <- colnames(biochem)[c(1, 6, 9, 14, 15, 24:28)]
biochem <- biochem[, keep]
# get weights for each individual mouse
# careful: did not come with column names
weight <- read_tsv(
"http://mtweb.cs.ucl.ac.uk/HSMICE/PHENOTYPES/weight",
col_names = F,
show_col_types = FALSE
)
# add column names
colnames(weight) <- c("subject_name", "weight")
# add weight to biochem table and get rid of NAs
# rename gender to sex
b <- inner_join(biochem, weight, by = "subject_name") |>
na.omit() |>
rename(sex = gender)Problem # 1
Can mouse sex explain mouse cholesterol? {.smaller}
STEP 1: Null hypothesis and variable specification — (2 pts)
\(\mathcal{H}_0:\) mouse \(sex\) does NOT explain \(cholesterol\)
\(cholesterol\) is the response variable
\(sex\) is the explanatory variable
STEP 2: Fit linear model and examine results — (3 pts)
fit_cs <- lm(formula = tot_cholesterol ~ 1 + sex, data = b)Fit summary:
glance(fit_cs) |>
gt() |>
fmt_number(columns = r.squared:statistic, decimals = 3)| r.squared | adj.r.squared | sigma | statistic | p.value | df | logLik | AIC | BIC | deviance | df.residual | nobs |
|---|---|---|---|---|---|---|---|---|---|---|---|
| 0.184 | 0.183 | 0.576 | 400.774 | 1.425116e-80 | 1 | -1546.04 | 3098.081 | 3114.537 | 591.5753 | 1780 | 1782 |
Coefficient summary:
tidy(fit_cs) |>
gt() |>
fmt_number(columns = estimate:statistic, decimals = 3)| term | estimate | std.error | statistic | p.value |
|---|---|---|---|---|
| (Intercept) | 2.797 | 0.019 | 144.812 | 0.000000e+00 |
| sexM | 0.547 | 0.027 | 20.019 | 1.425116e-80 |
Collecting residuals and other information — (2 pts)
add residuals and other information
STEP 4: Visualize the error around fit — (2 pts)
# plot of data with mean and colored by residuals
p_cs <- ggplot(
b_cs,
aes(x = sex, y = tot_cholesterol)
) +
geom_point(
position = position_jitter(),
aes(color = .resid)
) +
scale_color_gradient2(
low = "blue",
mid = "black",
high = "yellow"
) +
geom_hline(
yintercept = avg_c,
color = "darkgrey"
) +
geom_segment(
aes(
x = .5,
xend = 1.5,
y = avg_cf,
yend = avg_cf
),
color = "red"
) +
geom_segment(
aes(
x = 1.5,
xend = 2.5,
y = avg_cm
),
yend = avg_cm,
color = "red"
) +
theme_cowplot()
p_csWarning in geom_segment(aes(x = 0.5, xend = 1.5, y = avg_cf, yend = avg_cf), : All aesthetics have length 1, but the data has 1782 rows.
ℹ Please consider using `annotate()` or provide this layer with data containing
a single row.Warning in geom_segment(aes(x = 1.5, xend = 2.5, y = avg_cm), yend = avg_cm, : All aesthetics have length 1, but the data has 1782 rows.
ℹ Please consider using `annotate()` or provide this layer with data containing
a single row.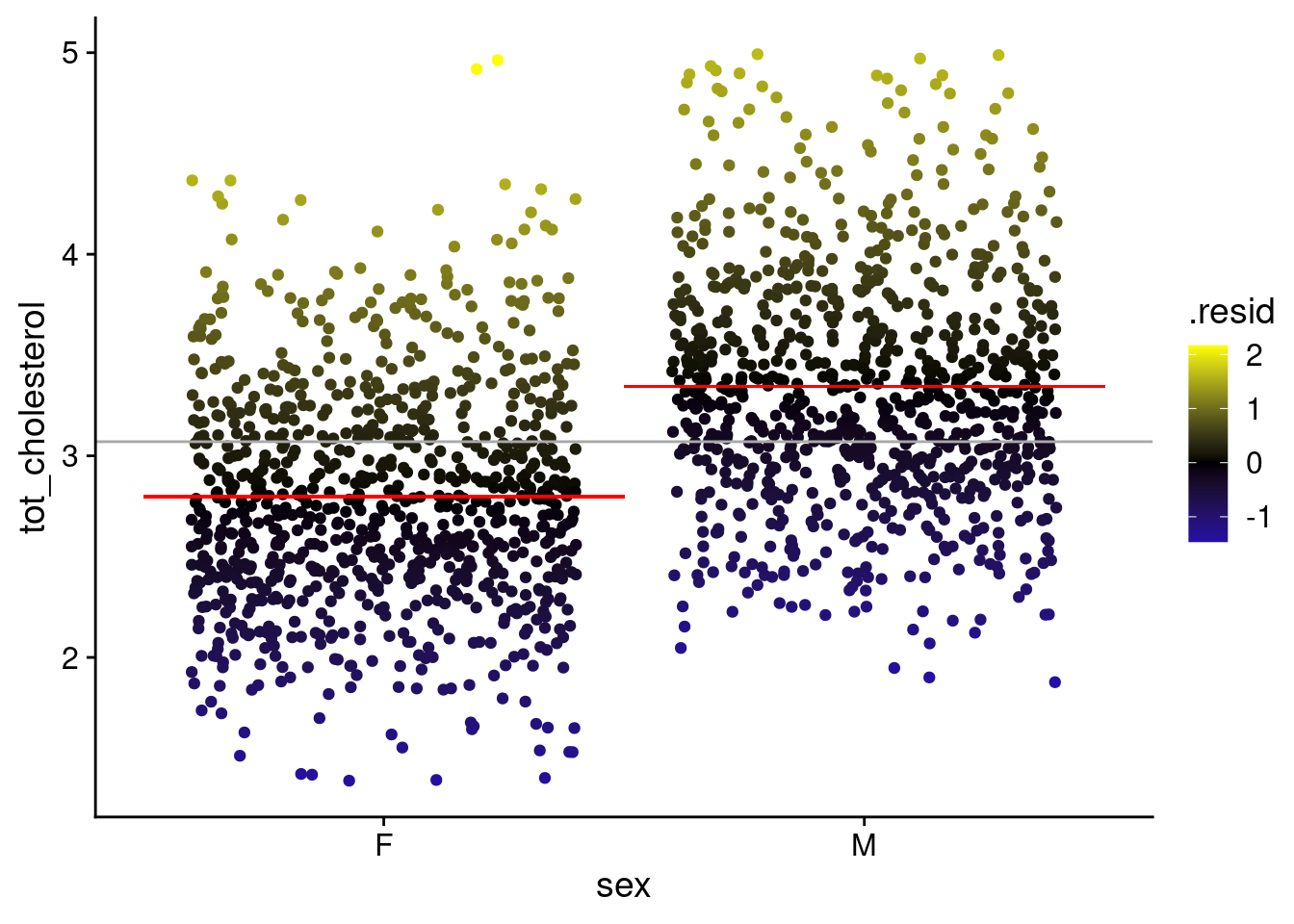
STEP 5: Visualize the error around the null (mean weight) — (2 pts)
p_c <- ggplot(
b_cs,
aes(x = sex, y = tot_cholesterol)
) +
geom_point(
position = position_jitter(),
aes(color = tot_cholesterol - avg_c)
) +
scale_color_gradient2(
low = "blue",
mid = "black",
high = "yellow"
) +
geom_hline(
yintercept = avg_c,
color = "darkgrey"
) +
theme_cowplot()
p_c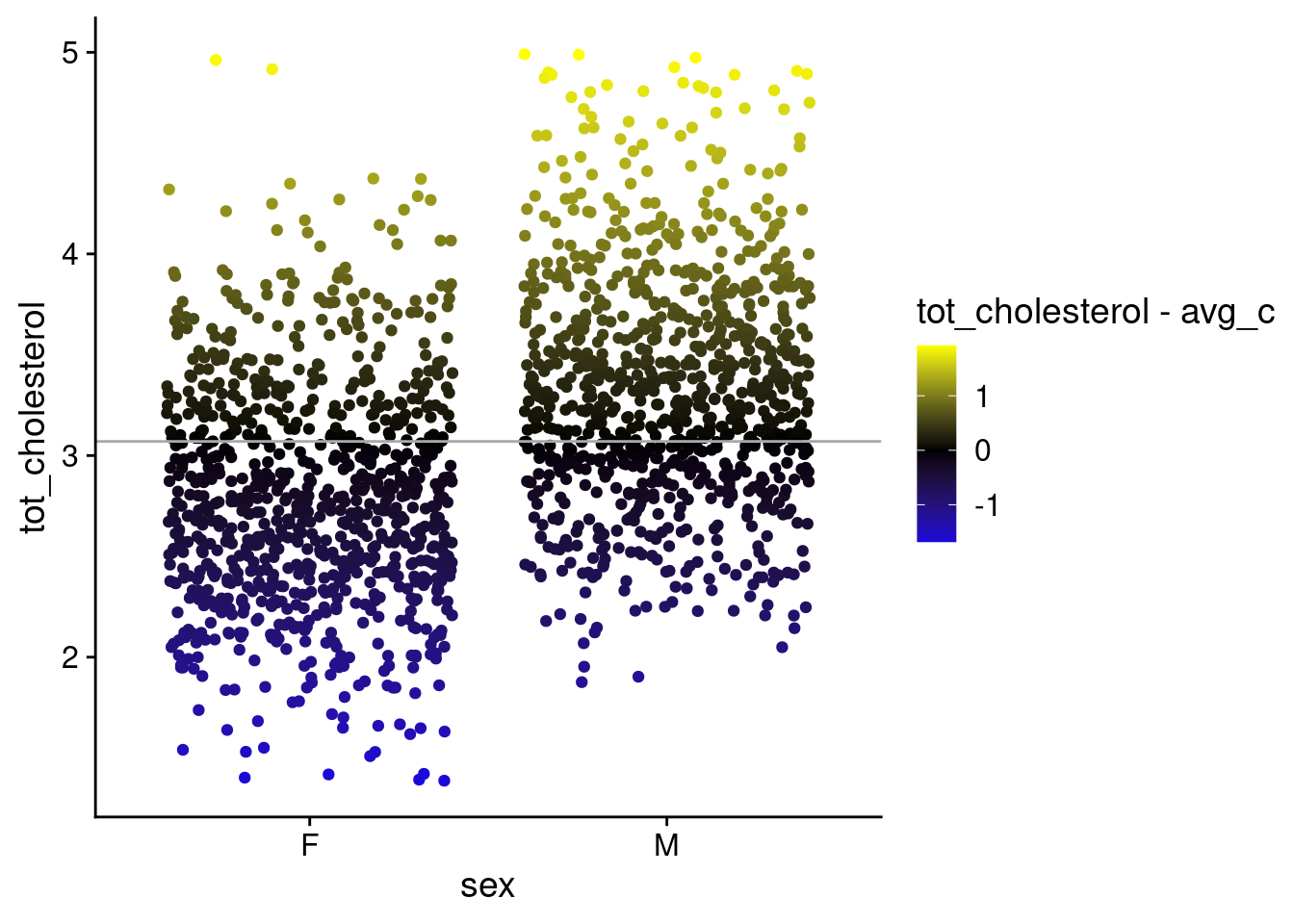
Plot the fit error and the null error as 2 panels — (2 pts)
Warning in geom_segment(aes(x = 0.5, xend = 1.5, y = avg_cf, yend = avg_cf), : All aesthetics have length 1, but the data has 1782 rows.
ℹ Please consider using `annotate()` or provide this layer with data containing
a single row.Warning in geom_segment(aes(x = 1.5, xend = 2.5, y = avg_cm), yend = avg_cm, : All aesthetics have length 1, but the data has 1782 rows.
ℹ Please consider using `annotate()` or provide this layer with data containing
a single row.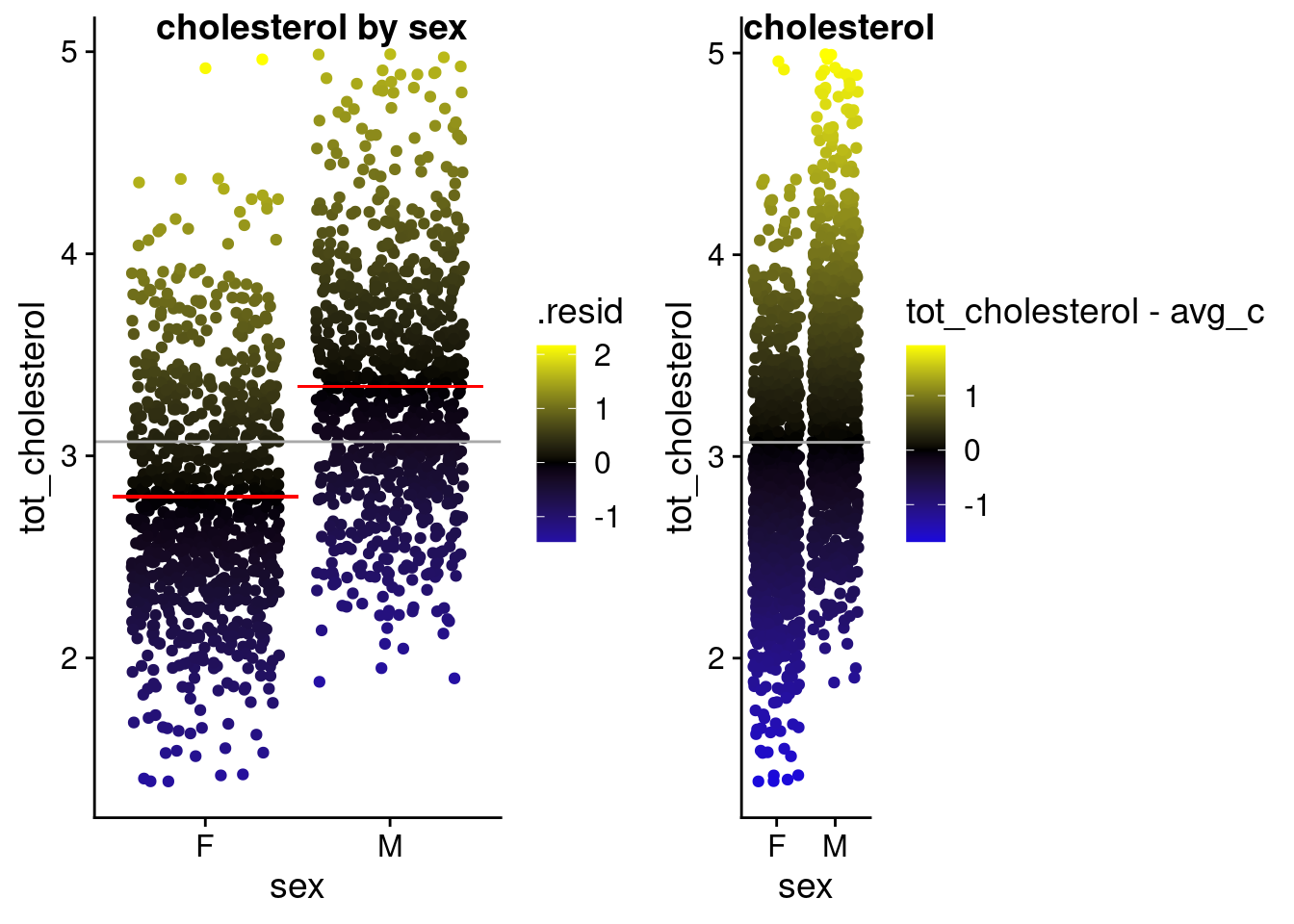
Calculate \(R^2\) — (3 pts)
\(R^2 = 1 - \displaystyle \frac {SS_{fit}}{SS_{null}}\)
rsq <- 1 - ss_fit / ss_null
rsq[1] 0.1837759check agains Rsq in your fit
Compare to traditional t-test — (2 pts)
| .y. | group1 | group2 | statistic | p |
|---|---|---|---|---|
| tot_cholesterol | F | M | -20.01933 | 1.46e-80 |
Provide your interpretation of the result — (2 pts)
The null model that mouse \(sex\) does NOT explain \(cholesterol\) is not well supported. Therefore, i believe that mouse \(sex\) is able to explain ~%18 of the variation in \(cholesterol\).