Use the data files in the data/ directory to answer the questions.
For this problem set, you are allowed to help each other, but you are not allowed to post correct answers in slack.
The problem set is due 12pm on Sept 11.
A quantitative PCR experiment
This problem set is a more complex version of the qPCR experiment we discussed in class.
Here is the experimental setup:
Three cell lines (wt, TF-mutant, RL-mutant) were treated with a drug that induces interferon expression.
After specific time points (0, 4, 8, 12, 24 hours), cells were harvested and actin and interferon mRNA were analyzed by quantitative PCR (with 3 biological replicates, and 3 technical replicates).
Data for the Biological replicates come from three independent cultures and RNA samples.
Data for the technical replicates come the same RNA sample, measured in 3 independent qPCR reactions.
-- Attaching core tidyverse packages ------------------------ tidyverse 2.0.0 --
v dplyr 1.1.3 v readr 2.1.4
v forcats 1.0.0 v stringr 1.5.0
v ggplot2 3.4.3 v tibble 3.2.1
v lubridate 1.9.2 v tidyr 1.3.0
v purrr 1.0.2
-- Conflicts ------------------------------------------ tidyverse_conflicts() --
x dplyr::filter() masks stats::filter()
x dplyr::lag() masks stats::lag()
i Use the conflicted package (<http://conflicted.r-lib.org/>) to force all conflicts to become errors
Rows: 18 Columns: 19
-- Column specification --------------------------------------------------------
Delimiter: "\t"
chr (19): row, 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18
i Use `spec()` to retrieve the full column specification for this data.
i Specify the column types or set `show_col_types = FALSE` to quiet this message.
Rows: 18 Columns: 19
-- Column specification --------------------------------------------------------
Delimiter: "\t"
chr (1): row
dbl (18): 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18
i Use `spec()` to retrieve the full column specification for this data.
i Specify the column types or set `show_col_types = FALSE` to quiet this message.
Tidy the data
Given the experimental setup and the shape of the tibbles, you should be able to answer: Are these data tidy?
What are the variables in the data?
genotype, time, gene, biological replicate, technical replicate
`summarise()` has grouped output by 'gt', 'time', 'gene'. You can override
using the `.groups` argument.
`summarise()` has grouped output by 'gt', 'time'. You can override using the
`.groups` argument.
Question 2
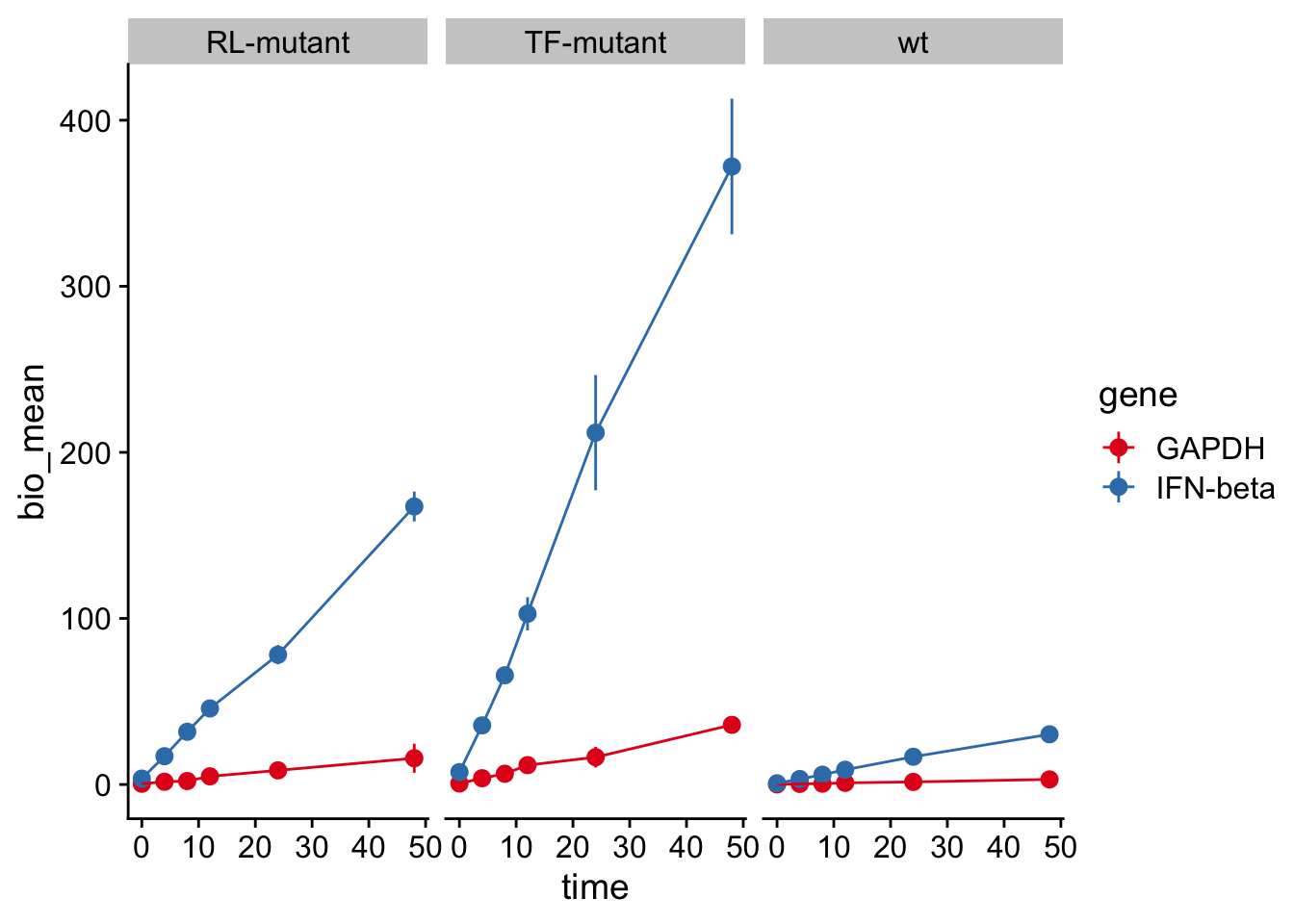
Create a plot of expression by time from the data, using the mean of the biological replicates as the y value.
What can you say about the expression of GAPDH and IFN in the different cell types?
Can you come up with a simple molecular mechanism to explain the results?
A straightforward explanation is that “TF” is a repssor of IFN induction (i.e., IFN goes up in its absence), and RL is similar, but possibly via an indirect effect on IFN expression. In any case, the mutants have negligible impact on GAPDH expression.
Question 3
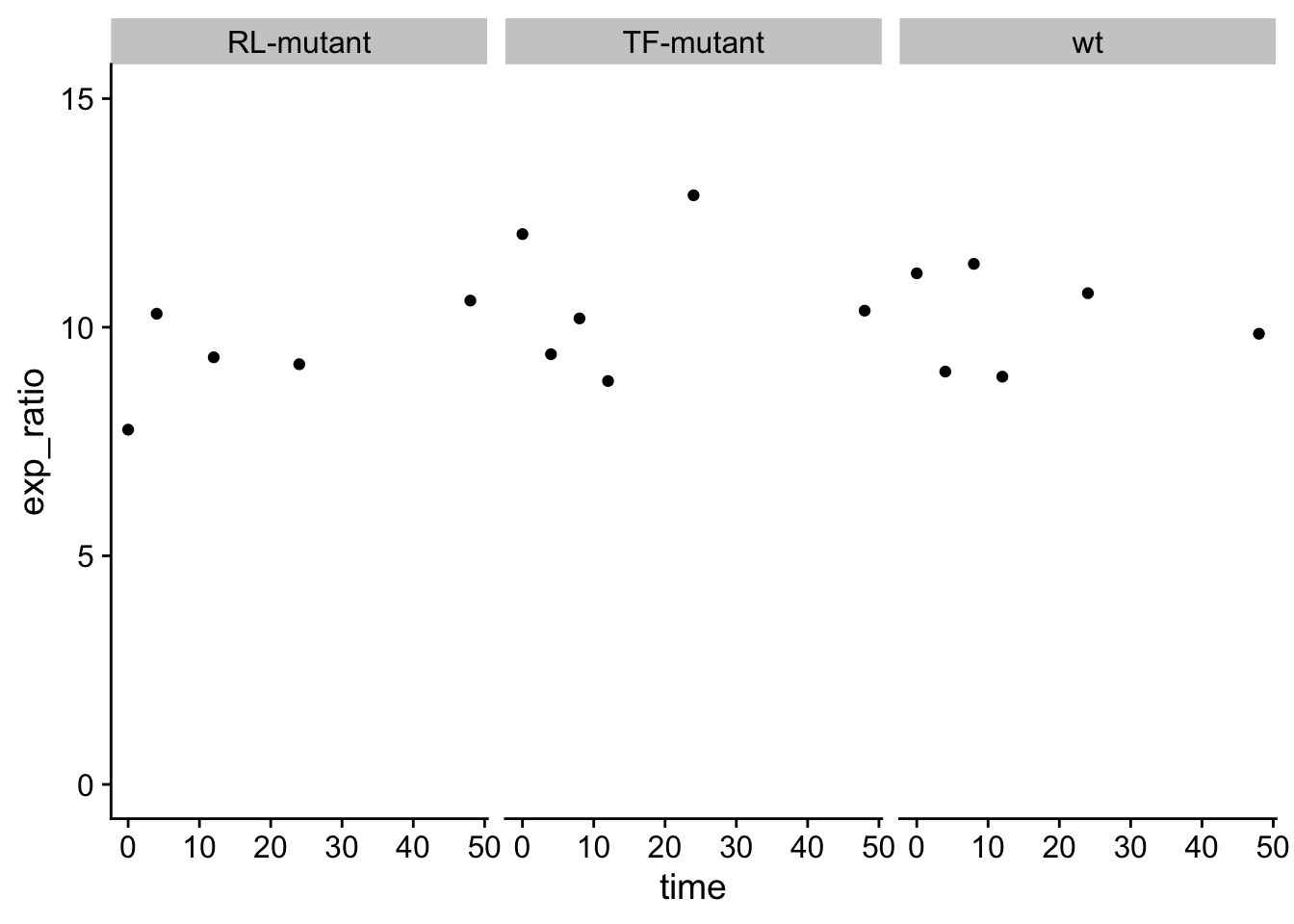
Reformat the data from question 2 such that you calculate a ratio of IFN to GAPDH. Start with the data question 1.2 above.
Re-plot the data as in question 2, but leave out the color as you have collapsed the two genes into one value.
This is considerably less interesting than I was planning, caused by the fractional values of GAPDH. C’est la vie. Consider this an exercise in pivoting.
Question 4
Is there greater variance across the technical replicates, or across the biological replicates (across the whole experiment)?
To get at this question, calculate the standard deviations across the two sets of replicates separately. Which one has a greater spread?
The key is the way you used group_by(). The technical replicates are tighter, which is expected if i) biology is variable and ii) you are any good at pipetting.