[,1] [,2] [,3] [,4] [,5]
[1,] 0 0 0 0 0
[2,] 0 1 0 0 0
[3,] 0 0 0 0 0
[4,] 2 0 0 0 0
[5,] 0 0 0 1 0Single cell RNA-seq class I
Overview
Today we will begin a two-course segment on single cell RNA-seq analysis. We will begin with an introduction to single cell RNA-seq data, followed by a discussion of key quality control metrics used to exclude low-quality cells from the data.
Analyzing single cell data in R
Droplet based scRNA-seq will produce libraries with 100,000 - 1,000,000 cell barcodes in each experiment, despite only 1-10,000 cells being loaded. Many of these barcodes are generated by non-cell containing empty droplets, which will end up with a few reads.
Storing a matrix of 20,000 genes x 1,000,000 cell barcodes would require a large amount of memory (20 billion values). However most of these values (> 95%) are zeros due to many empty barcodes with few UMIs and the low efficiency of single cell library generation (< 10-20% efficient). To limit unnecessary memory usage single cell data matrices are stored in a sparseMatrix format.
Converting a dense matrix to a sparse matrix can be accomplished using the generic as function (which is a common way to convert formats).
sm <- as(m, "sparseMatrix")
# alternatively
# Matrix(vals, nrow = 5)
sm5 x 5 sparse Matrix of class "dtCMatrix"
[1,] . . . . .
[2,] . 1 . . .
[3,] . . . . .
[4,] 2 . . . .
[5,] . . . 1 .The sparse matrix format only stores non-zero values in the matrix. Internally , and in text formats, these are stored as a row column value triplet.
summary(sm)5 x 5 sparse Matrix of class "dtCMatrix", with 3 entries
i j x
1 4 1 2
2 2 2 1
3 5 4 1Many of the functions that manipulate matrices (e.g. rowMeans, colSums, apply, [) can also be used on sparseMatrices, provided that you load the Matrix package (e.g library(Matrix)).
How can we extract the first 2 rows and first 3 columns of the sparse matrix sm that we generated above?
# print subset of sm
sm[1:2, 1:3]How can we calculate the sum of the columns of sm?
# find column sums of sparse
colSums(sm)Base R subsetting
To work with single cell data it will be helpful to know some base R concepts in such as subsetting with brackets [ and referencing and generating columns with $.
Vectors in R can be subset by vector index (position), a logical vector (c(TRUE, FALSE)), or name (if the vector is named).
Consider the following character vector letters.
letters [1] "a" "b" "c" "d" "e" "f" "g" "h" "i" "j" "k" "l" "m" "n" "o" "p" "q" "r" "s"
[20] "t" "u" "v" "w" "x" "y" "z"# extract 2nd, 4th, and 6th entry
letters[c(2, 4, 6)][1] "b" "d" "f"[1] "a" "e" "i" "o" "u" A Z
"a" "z" Matrices are 2 dimensional vectors and have similar subsetting rules except there are two dimensions, rows and columns.
matrix[rows_to_subset, columns_to_subset]
[,1] [,2] [,3] [,4]
[1,] 2 8 14 20
[2,] 4 10 16 22
[3,] 6 12 18 24# extract 2nd and 4th column
m[, c(2, 4)] [,1] [,2]
[1,] 7 19
[2,] 8 20
[3,] 9 21
[4,] 10 22
[5,] 11 23
[6,] 12 24# first 3 rows and 2nd and 4th column
m[1:3, c(2, 4)] [,1] [,2]
[1,] 7 19
[2,] 8 20
[3,] 9 21# extract rows with totals > 50
m[rowSums(m) > 50, ] [,1] [,2] [,3] [,4]
[1,] 4 10 16 22
[2,] 5 11 17 23
[3,] 6 12 18 24# extract columns with minimum values < 8
m[, colMins(m) < 8] [,1] [,2]
[1,] 1 7
[2,] 2 8
[3,] 3 9
[4,] 4 10
[5,] 5 11
[6,] 6 12The base R data.frame and Bioconductor DataFrame can also be subset with the [ and we can reference individual vectors in a data.frame using $.
# first 3 rows and columns of mtcars data.frame
mtcars[1:3, 1:3] mpg cyl disp
Mazda RX4 21.0 6 160
Mazda RX4 Wag 21.0 6 160
Datsun 710 22.8 4 108# columns can be referenced using $, which extracts a vector
mtcars$mpg [1] 21.0 21.0 22.8 21.4 18.7 18.1 14.3 24.4 22.8 19.2 17.8 16.4 17.3 15.2 10.4
[16] 10.4 14.7 32.4 30.4 33.9 21.5 15.5 15.2 13.3 19.2 27.3 26.0 30.4 15.8 19.7
[31] 15.0 21.4# columns can be generated or overwritten using $ with assignment
mtcars$new_column_name <- "Hello!"
mtcars$wt <- mtcars$wt * 1000
# We can subset using logical vectors
# E.g. filter for rows (cars) with mpg > 20
mtcars[mtcars$mpg > 20, ] mpg cyl disp hp drat wt qsec vs am gear carb
Mazda RX4 21.0 6 160.0 110 3.90 2620 16.46 0 1 4 4
Mazda RX4 Wag 21.0 6 160.0 110 3.90 2875 17.02 0 1 4 4
Datsun 710 22.8 4 108.0 93 3.85 2320 18.61 1 1 4 1
Hornet 4 Drive 21.4 6 258.0 110 3.08 3215 19.44 1 0 3 1
Merc 240D 24.4 4 146.7 62 3.69 3190 20.00 1 0 4 2
Merc 230 22.8 4 140.8 95 3.92 3150 22.90 1 0 4 2
Fiat 128 32.4 4 78.7 66 4.08 2200 19.47 1 1 4 1
Honda Civic 30.4 4 75.7 52 4.93 1615 18.52 1 1 4 2
Toyota Corolla 33.9 4 71.1 65 4.22 1835 19.90 1 1 4 1
Toyota Corona 21.5 4 120.1 97 3.70 2465 20.01 1 0 3 1
Fiat X1-9 27.3 4 79.0 66 4.08 1935 18.90 1 1 4 1
Porsche 914-2 26.0 4 120.3 91 4.43 2140 16.70 0 1 5 2
Lotus Europa 30.4 4 95.1 113 3.77 1513 16.90 1 1 5 2
Volvo 142E 21.4 4 121.0 109 4.11 2780 18.60 1 1 4 2
new_column_name
Mazda RX4 Hello!
Mazda RX4 Wag Hello!
Datsun 710 Hello!
Hornet 4 Drive Hello!
Merc 240D Hello!
Merc 230 Hello!
Fiat 128 Hello!
Honda Civic Hello!
Toyota Corolla Hello!
Toyota Corona Hello!
Fiat X1-9 Hello!
Porsche 914-2 Hello!
Lotus Europa Hello!
Volvo 142E Hello!Reading alevin output into R with tximport
Our old-friend, the tximport package, has methods for importing the binary data from alevin. We need to supply a path to the quants_mat.gz file. Note that in contrast to importing data from salmon, tximport only allows 1 file to be loaded. If you want to load multiple samples use iteration approaches (e.g. lapply, purrr::map, a for loop). Also note that the eds package was installed which greatly speeds up the loading of the matrix.
We will load in data from a 10x Genomics scRNA-seq library generated from human periperhal blood mononuclear cells (PMBCS).
importing alevin data is much faster after installing 'eds'reading in alevin gene-level counts across cells names(tx)[1] "abundance" "counts" "countsFromAbundance"tx is a list with 3 elements, abundance, counts, and countsFromAbundance. Let’s look at the counts element
mat <- tx$counts
mat[5:10, 1:3]6 x 3 sparse Matrix of class "dgTMatrix"
GCTGCAGTCCGATCTC ACTATGGAGGTCCCTG ATTTCTGTCTCTATGT
ENSG00000243485 . . .
ENSG00000284332 . . .
ENSG00000237613 . . .
ENSG00000268020 . . .
ENSG00000290826 . . .
ENSG00000240361 . . .Here you can see that tx$counts is a sparse matrix that is genes (rows) by cells (columns).
How many barcodes are in tx$counts? How many genes?
# TODO Find number of barcodes and genes in tx$counts
dim(mat)What fraction of the matrix is non-zero? We can use the nnzerofunction from the Matrix package check
The SingleCellExperiment class
Single cell analysis is centered around the SingleCellExperiment data structure, which serves as a single container to store the input data, various transformations (count data, normalized count data, PCA, UMAPs, etc.) and any results that are generated.
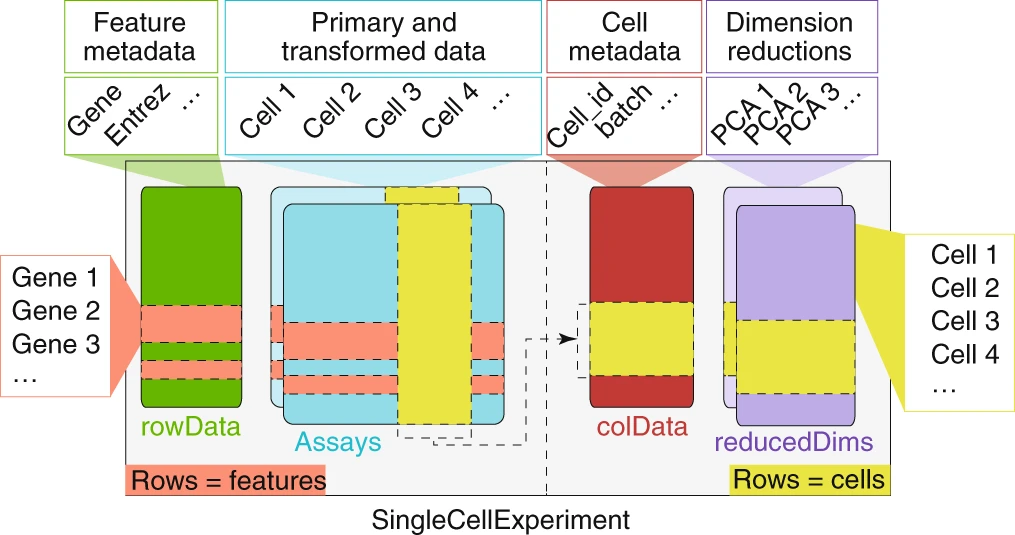
Creating a SingleCellExperiment object
A SingleCellExperiment object can be created from our sparse matrix using the SingleCellExperiment() function.
sce <- SingleCellExperiment(list(counts = mat))
sceclass: SingleCellExperiment
dim: 62266 6075
metadata(0):
assays(1): counts
rownames(62266): ENSG00000290825 ENSG00000223972 ... ENSG00000210195
ENSG00000210196
rowData names(0):
colnames(6075): GCTGCAGTCCGATCTC ACTATGGAGGTCCCTG ... ACGTAGGGTGACAGCA
TCTCAGCTCGCCGAAC
colData names(0):
reducedDimNames(0):
mainExpName: NULL
altExpNames(0):The SingleCellExperiment object stores the gene x cell count matrix within assays().
# get list of assays
assays(sce)List of length 1
names(1): counts# extract single assay
assay(sce, "counts")[1:4, 1:4]4 x 4 sparse Matrix of class "dgTMatrix"
GCTGCAGTCCGATCTC ACTATGGAGGTCCCTG ATTTCTGTCTCTATGT
ENSG00000290825 . . .
ENSG00000223972 . . .
ENSG00000227232 . . .
ENSG00000278267 . . .
TATCTGTAGGTGATAT
ENSG00000290825 .
ENSG00000223972 .
ENSG00000227232 .
ENSG00000278267 .assays(sce)$counts[1:4, 1:4]4 x 4 sparse Matrix of class "dgTMatrix"
GCTGCAGTCCGATCTC ACTATGGAGGTCCCTG ATTTCTGTCTCTATGT
ENSG00000290825 . . .
ENSG00000223972 . . .
ENSG00000227232 . . .
ENSG00000278267 . . .
TATCTGTAGGTGATAT
ENSG00000290825 .
ENSG00000223972 .
ENSG00000227232 .
ENSG00000278267 .# convenience function for counts assay
counts(sce)[1:4, 1:4]4 x 4 sparse Matrix of class "dgTMatrix"
GCTGCAGTCCGATCTC ACTATGGAGGTCCCTG ATTTCTGTCTCTATGT
ENSG00000290825 . . .
ENSG00000223972 . . .
ENSG00000227232 . . .
ENSG00000278267 . . .
TATCTGTAGGTGATAT
ENSG00000290825 .
ENSG00000223972 .
ENSG00000227232 .
ENSG00000278267 .Accessing and storing cell and gene level metadata
As we perform analyses we will accumulate cell-level information, such as quality control metrics, clustering results, and celltype assignments. The SingleCellExperiment class stores this data within a data frame called colData, which we can access using colData(). This is a specialized Bioconductor specific data.frame class (DataFrame) which has similar semantics and functionality to a base R data.frame.
# empty right now.
colData(sce)DataFrame with 6075 rows and 0 columns# add a sample annotation
colData(sce)$cell_source <- "PBMC"
# equivalent approach using $
sce$cell_source <- "PBMC"
colData(sce)DataFrame with 6075 rows and 1 column
cell_source
<character>
GCTGCAGTCCGATCTC PBMC
ACTATGGAGGTCCCTG PBMC
ATTTCTGTCTCTATGT PBMC
TATCTGTAGGTGATAT PBMC
AGCCAGCCAAAGCACG PBMC
... ...
CATCCCAAGTACTCGT PBMC
CTCCTCCCATGAAGCG PBMC
AGTTCCCCATGTCAGT PBMC
ACGTAGGGTGACAGCA PBMC
TCTCAGCTCGCCGAAC PBMCThe SingleCellExperiment also stores gene-level metadata in a data.frame called rowData(). We will use the rowData to store gene ids, symbols, and other information about genes.
# empty right now
rowData(sce)DataFrame with 62266 rows and 0 columnsManipulating and subsetting a SingleCellExperiment
To get familiar with SingleCellExperiment objects let’s calculate the total number of counts in each cell and store these counts in the `colData().
sce$total_counts <- colSums(counts(sce))Next let’s calculate the total number of counts for each gene, summed across cells and the number of cells with > 0 counts per gene, and store both of these values in the rowData().
DataFrame with 62266 rows and 2 columns
total_gene_counts n_cells_expr
<numeric> <integer>
ENSG00000290825 65.594607 43
ENSG00000223972 0.000000 0
ENSG00000227232 5.506349 24
ENSG00000278267 0.000000 0
ENSG00000243485 0.333333 1
... ... ...
ENSG00000198695 2054 1509
ENSG00000210194 0 0
ENSG00000198727 274421 5704
ENSG00000210195 0 0
ENSG00000210196 0 0We can subset the SingleCellExperiment using the same syntax used to subset base R data.frames and matrices. Note that dplyr verbs do not work with SingleCellExperiment (see alternative tidySingleCellExperiment).
data.frame[rows, columns]
sce[genes, cells]# subset to data from first 4 genes and cells
sce[1:4, 1:4]
# subset to cells from PBMC cells
sce[, sce$cell_source == "PBMC"]
genes_to_keep <- c("ENSG00000223972", "ENSG00000210195", "ENSG00000210196")
sce[genes_to_keep, ]
cells_to_keep <- c("ACTATGGAGGTCCCTG", "GCTGCAGTCCGATCTC", "TCTCAGCTCGCCGAAC")
sce[, cells_to_keep]Many additional functions used on data.frames also work on SingleCellExperiment:
ncol(): # of cellsnrow(): # of genedims(): # of genes and cellsrownames(): rownames in matrices (e.g. genes)colnames(): colnames in matrices (e.g. cells)cbind(): combine multiple SingleCellExperiments by columnrbind(): combine multiple SingleCellExperiments by row
Storing gene identifier information
You’ll notice that our matrix has ensembl gene ids as the rownames (e.g. ENSG00000289576, ENSG00000221539). This is intentional as these identifiers are guaranteed to be unique and are a more stable and reliable identifier than gene symbols (e.g. ACTB, GAPDH). This becomes important if you want to compare to external datasets or ensure that your data can be easily used by others in the future.
These identifiers are useful but not as easy to interpret as gene symbols. Next we will retrieve gene symbols from an AnnotationHub() ensembldb resource and store the identifiers in the rowData().
ah <- AnnotationHub()
# download ensembl database
ens_db <- ah[["AH113665"]]loading from cacheDataFrame with 62266 rows and 4 columns
total_gene_counts n_cells_expr gene gene_id
<numeric> <integer> <character> <character>
ENSG00000290825 65.594607 43 DDX11L2 ENSG00000290825
ENSG00000223972 0.000000 0 DDX11L1 ENSG00000223972
ENSG00000227232 5.506349 24 WASH7P ENSG00000227232
ENSG00000278267 0.000000 0 MIR6859-1 ENSG00000278267
ENSG00000243485 0.333333 1 MIR1302-2HG ENSG00000243485
... ... ... ... ...
ENSG00000198695 2054 1509 MT-ND6 ENSG00000198695
ENSG00000210194 0 0 MT-TE ENSG00000210194
ENSG00000198727 274421 5704 MT-CYB ENSG00000198727
ENSG00000210195 0 0 MT-TT ENSG00000210195
ENSG00000210196 0 0 MT-TP ENSG00000210196We’d like to set the rownames of the object to symbols, but some of these are , NA or duplicated which will cause problems. uniquifyFeatureNames() is a convenience function that will rename gene symbols that are NA or duplicated values to the ensembl ID or a combination of gene symbol and ensembl ID.
[1] "DDX11L2_ENSG00000290825" "DDX11L1"
[3] "WASH7P" "MIR6859-1"
[5] "MIR1302-2HG" "MIR1302-2" Quality control and cell filtering
Now that we have our data in a SingleCellExperiment we will perform some filtering and quality control to remove low expression genes and poor quality cells.
Our SingleCellExperiment has 62266 genes in the matrix. Most of these are not expressed. We want to exclude these genes as they won’t provide any useful data for the analysis.
# exclude genes expressed in fewer than 10 cells (~ 1% of cells)
rowData(sce)$n_cells <- rowSums(counts(sce) > 0)
sce <- sce[rowData(sce)$n_cells >= 10, ]
sceclass: SingleCellExperiment
dim: 20858 6075
metadata(0):
assays(1): counts
rownames(20858): DDX11L2_ENSG00000290825 WASH7P ... MT-ND6 MT-CYB
rowData names(5): total_gene_counts n_cells_expr gene gene_id n_cells
colnames(6075): GCTGCAGTCCGATCTC ACTATGGAGGTCCCTG ... ACGTAGGGTGACAGCA
TCTCAGCTCGCCGAAC
colData names(2): cell_source total_counts
reducedDimNames(0):
mainExpName: NULL
altExpNames(0):To exclude low-quality cells we will use the following metrics:
- Number of counts per cell barcode
- Number of genes detected per barcode
- The percentage of counts from mitochondrial genes per barcode
A low number of counts, a low number of detected genes, and a high percentage of mitochondrial counts suggests that the cell had a broken membrane and the cytoplasmic mRNA leaked out. Conversely, an abnormally high number of counts and detected genes could indicate the presence of a doublet. See publication for more info (Classification of low quality cells from single-cell RNA-seq data)
To calculate these metrics we can use addPerCellQCMetrics from scater. Mitochondrial genes are named with a common “MT-” prefix (e.g. MT-CO2, MT-ATP6, MR-RNR2), which we can use to identify them.
# identify subset of genes that are from mitochondrial genome
is_mito <- startsWith(rowData(sce)$gene, "MT-")
sce <- addPerCellQCMetrics(sce, subsets = list(Mito = is_mito))Warning in .pause_resource(): omp_pause_resource() failed to relinquish
resources on device 0
Warning in .pause_resource(): omp_pause_resource() failed to relinquish
resources on device 0
Warning in .pause_resource(): omp_pause_resource() failed to relinquish
resources on device 0
Warning in .pause_resource(): omp_pause_resource() failed to relinquish
resources on device 0
Warning in .pause_resource(): omp_pause_resource() failed to relinquish
resources on device 0
Warning in .pause_resource(): omp_pause_resource() failed to relinquish
resources on device 0
Warning in .pause_resource(): omp_pause_resource() failed to relinquish
resources on device 0
Warning in .pause_resource(): omp_pause_resource() failed to relinquish
resources on device 0
Warning in .pause_resource(): omp_pause_resource() failed to relinquish
resources on device 0
Warning in .pause_resource(): omp_pause_resource() failed to relinquish
resources on device 0
Warning in .pause_resource(): omp_pause_resource() failed to relinquish
resources on device 0
Warning in .pause_resource(): omp_pause_resource() failed to relinquish
resources on device 0
Warning in .pause_resource(): omp_pause_resource() failed to relinquish
resources on device 0
Warning in .pause_resource(): omp_pause_resource() failed to relinquish
resources on device 0
Warning in .pause_resource(): omp_pause_resource() failed to relinquish
resources on device 0
Warning in .pause_resource(): omp_pause_resource() failed to relinquish
resources on device 0
Warning in .pause_resource(): omp_pause_resource() failed to relinquish
resources on device 0
Warning in .pause_resource(): omp_pause_resource() failed to relinquish
resources on device 0
Warning in .pause_resource(): omp_pause_resource() failed to relinquish
resources on device 0
Warning in .pause_resource(): omp_pause_resource() failed to relinquish
resources on device 0colData(sce)DataFrame with 6075 rows and 8 columns
cell_source total_counts sum detected subsets_Mito_sum
<character> <numeric> <numeric> <integer> <numeric>
GCTGCAGTCCGATCTC PBMC 31924 31908.0 5718 2668.49
ACTATGGAGGTCCCTG PBMC 35845 35801.5 6073 3963.23
ATTTCTGTCTCTATGT PBMC 31788 31758.3 6377 2496.33
TATCTGTAGGTGATAT PBMC 32025 31998.0 6260 2961.15
AGCCAGCCAAAGCACG PBMC 29882 29856.0 5746 2660.44
... ... ... ... ... ...
CATCCCAAGTACTCGT PBMC 151 151.000 132 12
CTCCTCCCATGAAGCG PBMC 133 132.500 131 6
AGTTCCCCATGTCAGT PBMC 173 172.667 166 8
ACGTAGGGTGACAGCA PBMC 255 253.000 217 13
TCTCAGCTCGCCGAAC PBMC 144 144.000 149 4
subsets_Mito_detected subsets_Mito_percent total
<integer> <numeric> <numeric>
GCTGCAGTCCGATCTC 13 8.36307 31908.0
ACTATGGAGGTCCCTG 15 11.07002 35801.5
ATTTCTGTCTCTATGT 15 7.86038 31758.3
TATCTGTAGGTGATAT 15 9.25417 31998.0
AGCCAGCCAAAGCACG 15 8.91092 29856.0
... ... ... ...
CATCCCAAGTACTCGT 5 7.94702 151.000
CTCCTCCCATGAAGCG 5 4.52830 132.500
AGTTCCCCATGTCAGT 6 4.63320 172.667
ACGTAGGGTGACAGCA 6 5.13834 253.000
TCTCAGCTCGCCGAAC 2 2.77778 144.000We can use the plotColData() function from scater to plot various metrics (as a ggplot2 object).
plotColData(sce, y = "sum")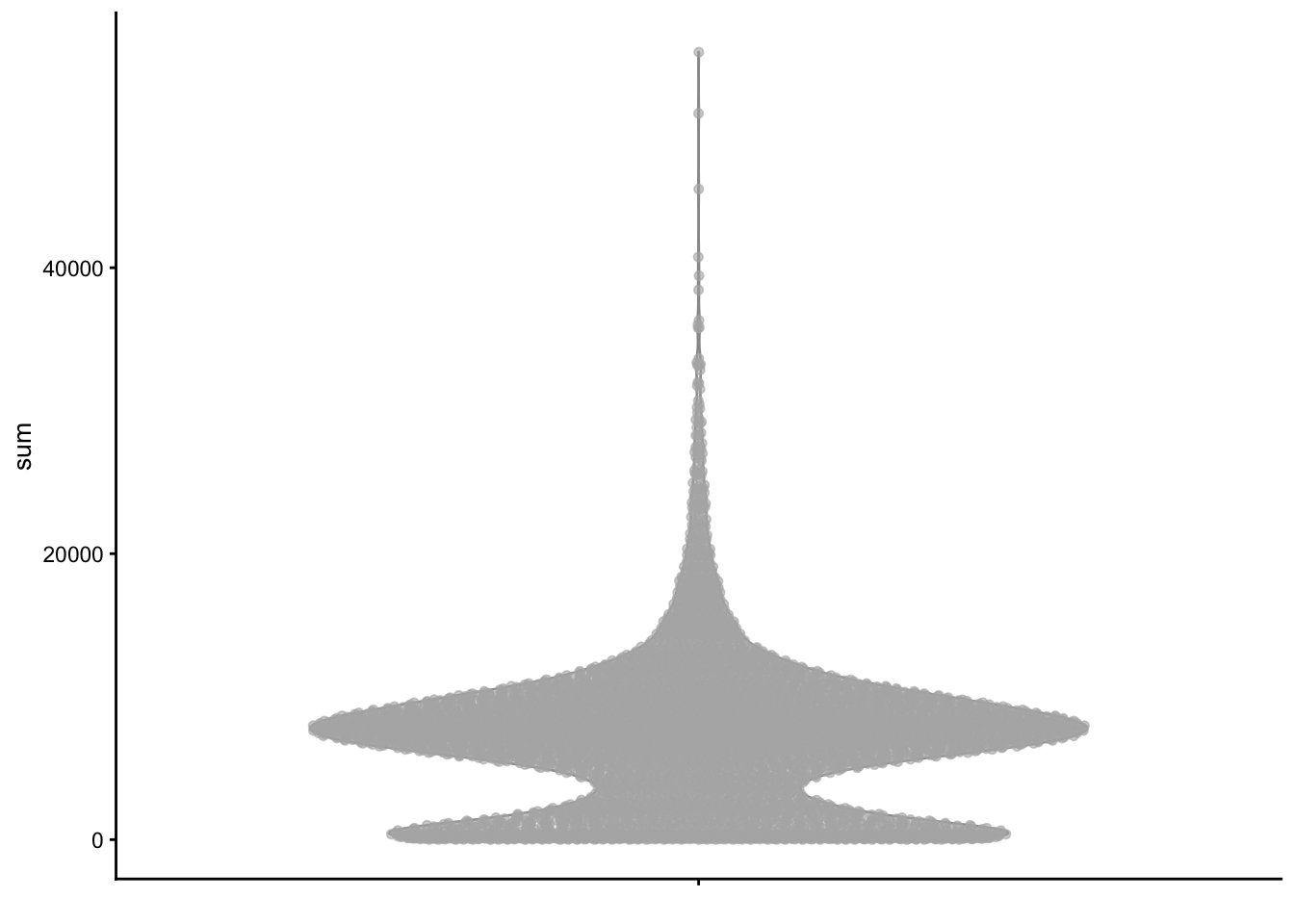
plotColData(sce, y = "detected")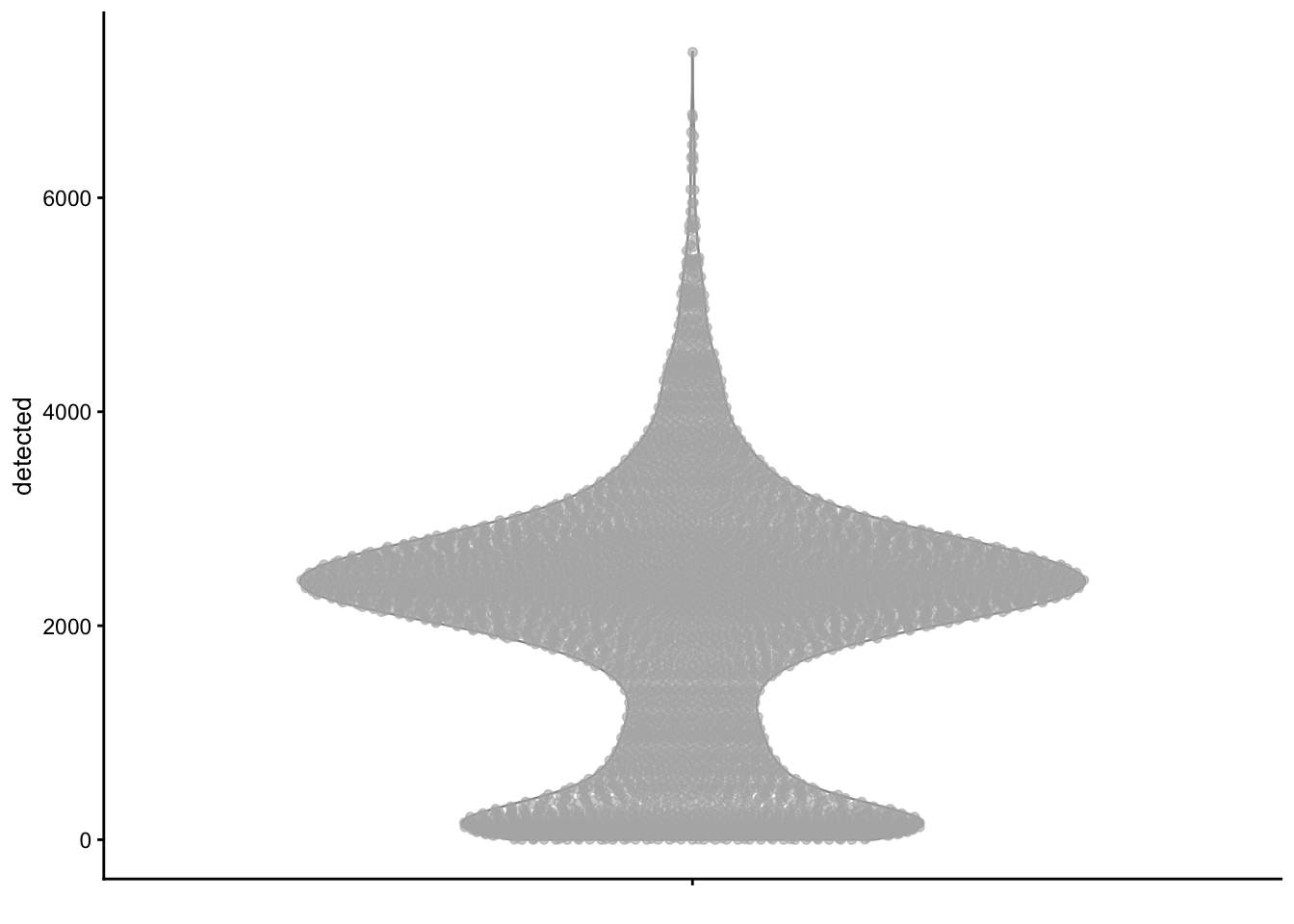
plotColData(sce, y = "detected", x = "sum", colour_by = "subsets_Mito_percent")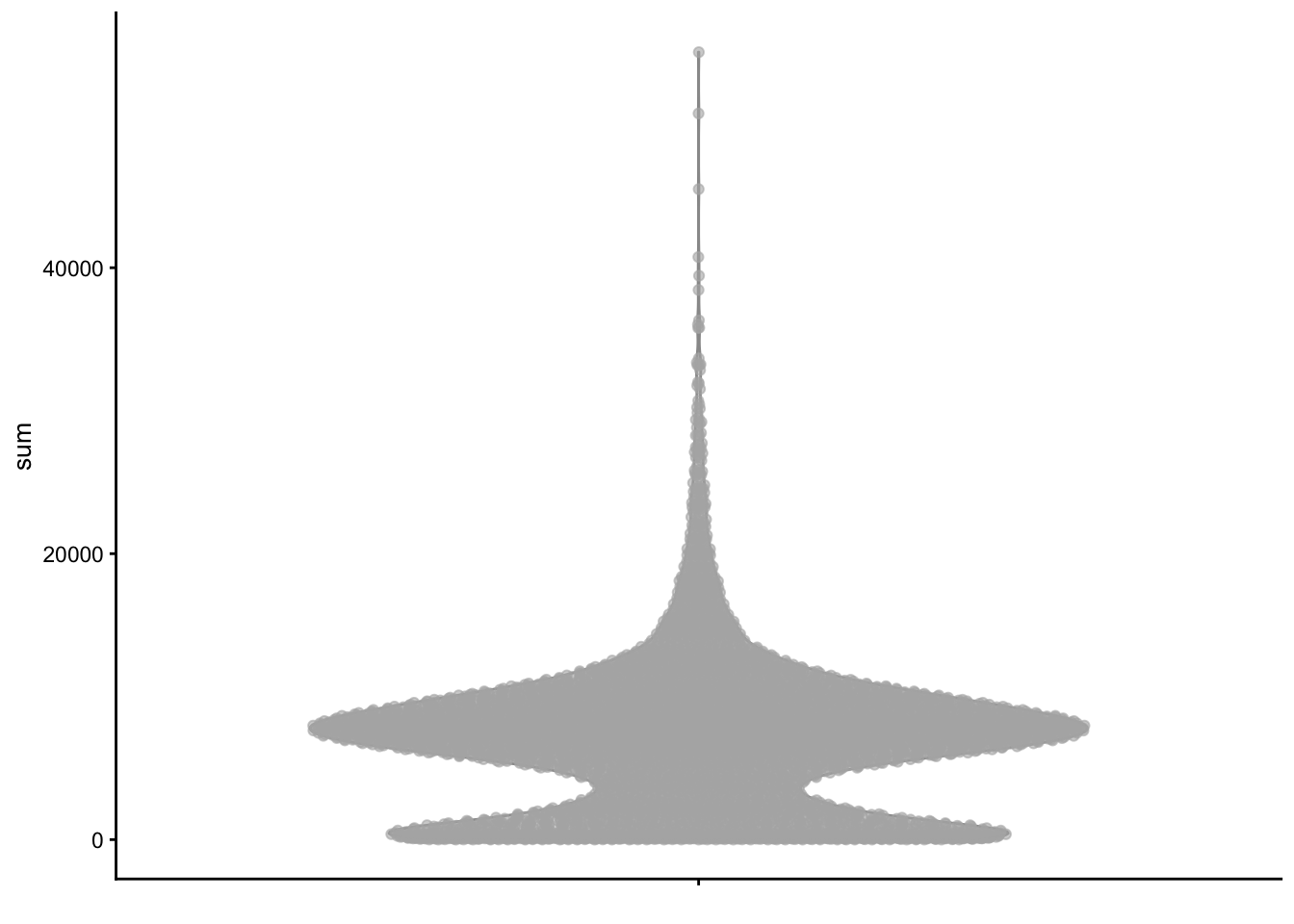
plotColData(sce, y = "subsets_Mito_percent", x = "sum") +
labs(x = "# of counts")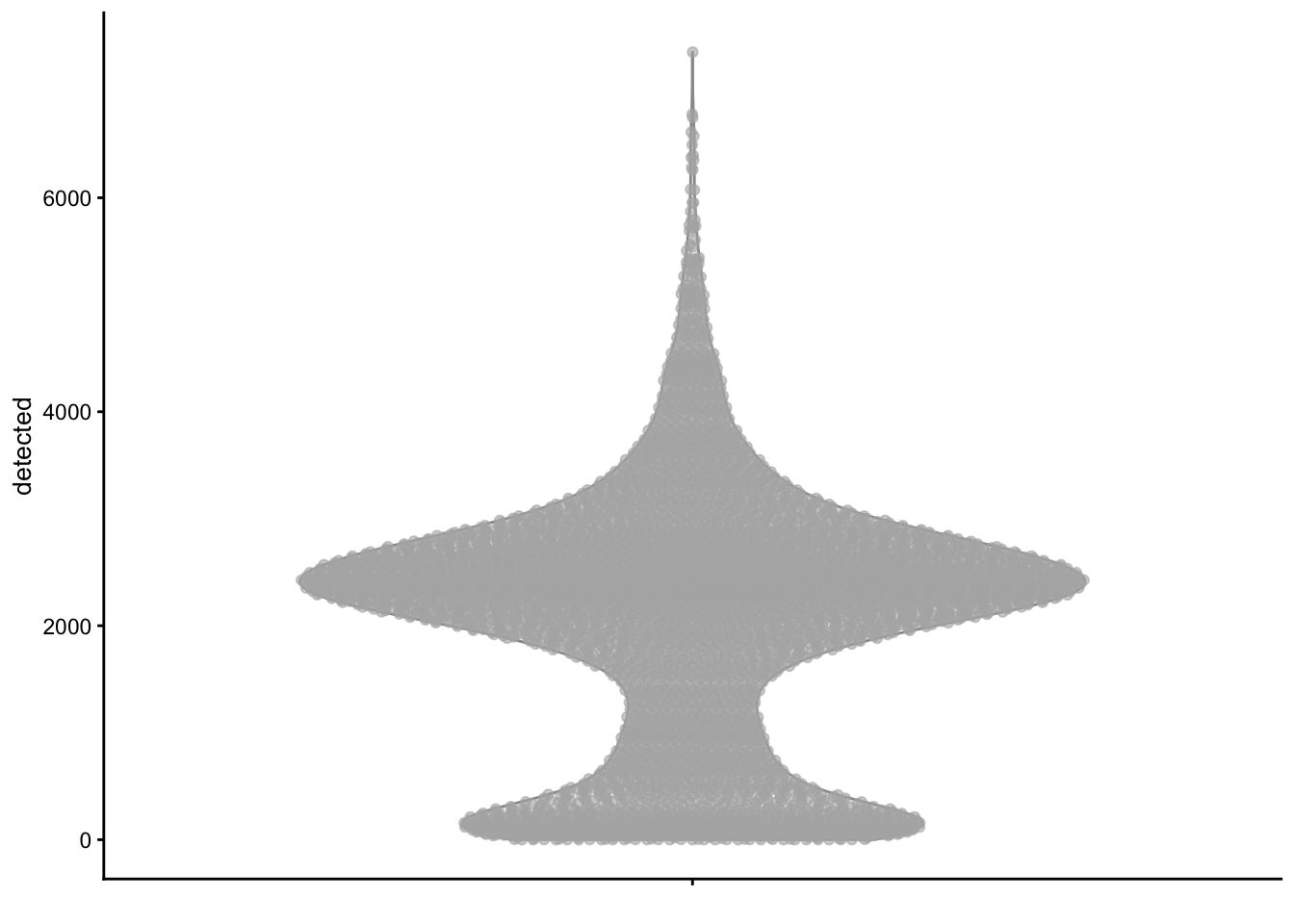
Also note that you can easily extract the colData() as a data.frame to use with ggplot2 for custom plots. To extract additional features, e.g. per cell gene expression, you can use makePerCellDF() or makePerFeatureDF() from scater.
cell_info <- as.data.frame(colData(sce))
ggplot(cell_info, aes(sum, subsets_Mito_percent)) +
geom_point()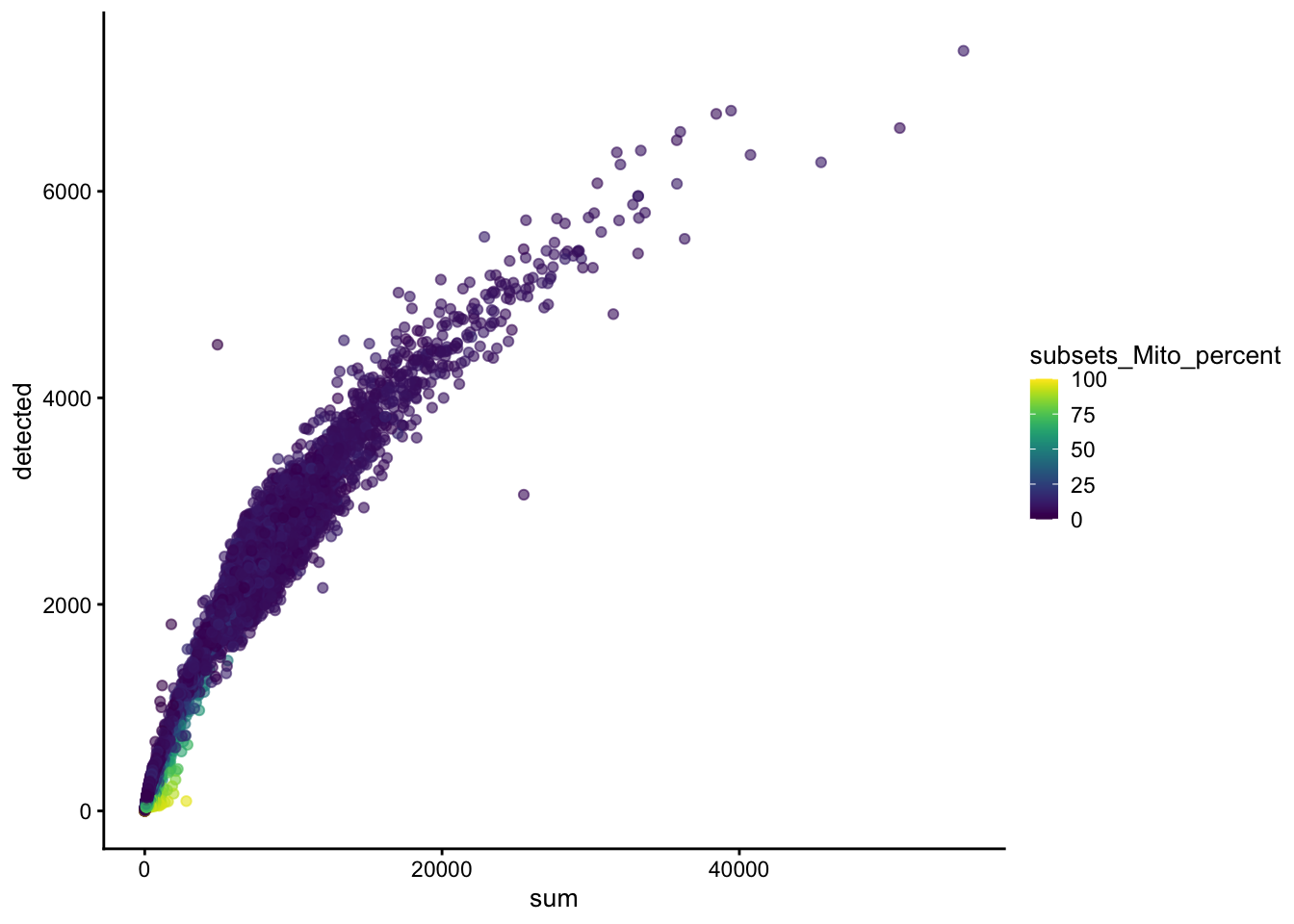
Selecting an appropriate cutoff can be somewhat arbitrary, and there is a risk of excluding meaningful cell populations. I suggest starting with lenient cutoffs, then later increasing the stringency after examining the clustering and cell types.
Let’s define high quality cell as those with less than 20% counts from mitocondrial RNAs, greater than 500 genes detected, and greater than 1000 total counts. How many cells pass these criteria?
pass_qc <- sce$subsets_Mito_percent < 20 & sce$detected > 500 & sce$sum > 1000
sum(pass_qc)[1] 4565Labeling the qc failed cells in plots can be helpful.
sce$pass_qc <- sce$subsets_Mito_percent < 20 &
sce$detected > 500 &
sce$sum > 1000
plotColData(sce, y = "subsets_Mito_percent", x = "sum", colour_by = "pass_qc") +
labs(x = "# of counts")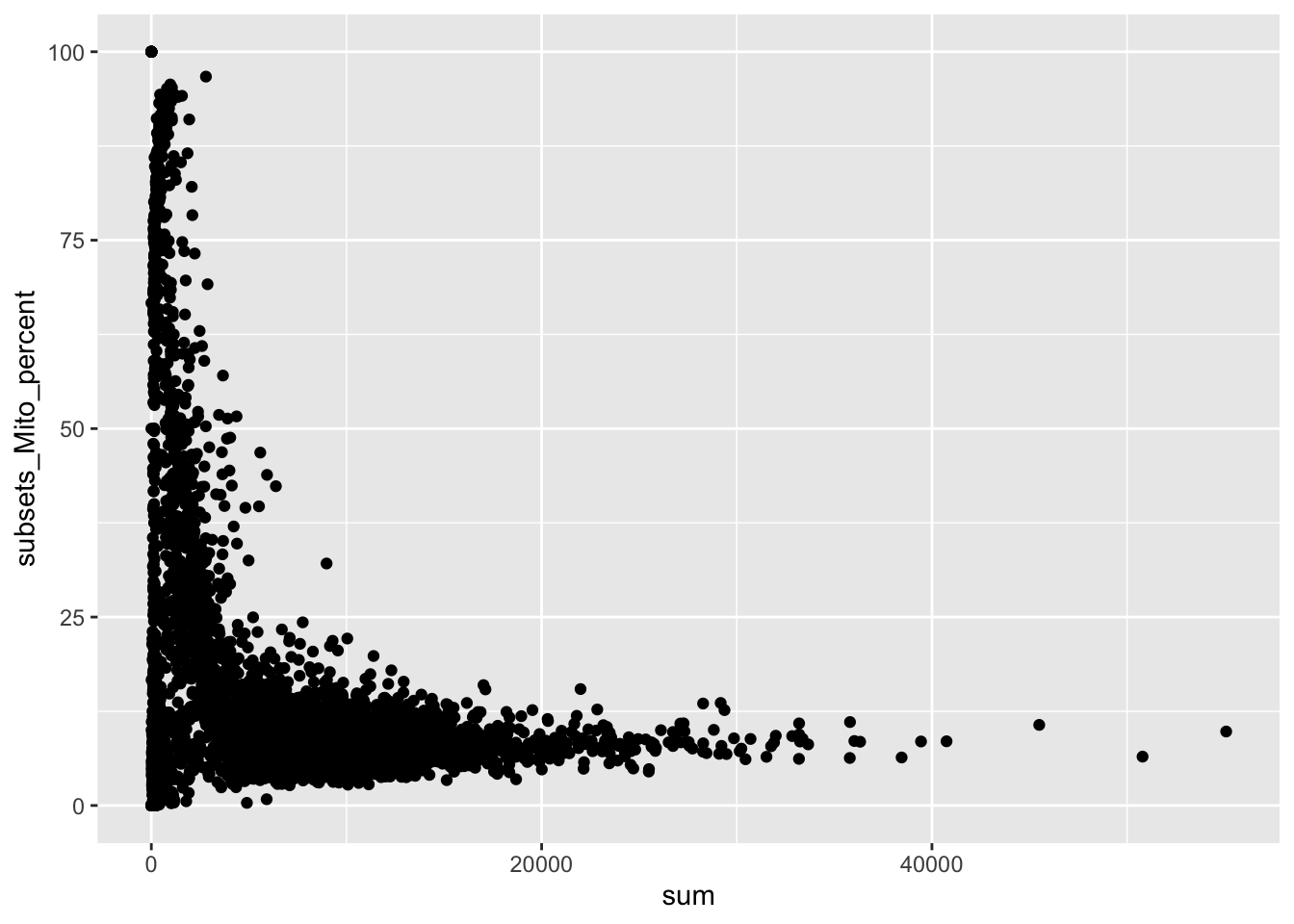
Lastly we can subset the SingleCellExperiment to exclude the low-quality cells.
sce <- sce[, sce$pass_qc]
sceclass: SingleCellExperiment
dim: 20858 4565
metadata(0):
assays(1): counts
rownames(20858): DDX11L2_ENSG00000290825 WASH7P ... MT-ND6 MT-CYB
rowData names(6): total_gene_counts n_cells_expr ... n_cells
subsets_Mito
colnames(4565): GCTGCAGTCCGATCTC ACTATGGAGGTCCCTG ... CTGGATCCACCGTACG
TACAACGGTCTCGCGC
colData names(9): cell_source total_counts ... total pass_qc
reducedDimNames(0):
mainExpName: NULL
altExpNames(0):It is often a good idea to save the SingleCellExperiment at periodic steps in the analysis. Use saveRDS() to store the object as a file, which can later be imported back into R using readRDS().
saveRDS(sce, "path/to/a/file.rds")Analysis steps revisited
Normalization
Normalization attempts to correct for technical biases that will distort biological signal in the data. A large source of variation arises due to differences in sequencing depth between cells. This can be seen by performing PCA on the unnormalized counts. We will use runPCA from scater to perform PCA.
# set seed for functions with a randomized component
# to obtain the same result each execution
set.seed(20231023)
sce <- runPCA(sce, exprs_values = "counts", name = "count_PCA")Warning in .pause_resource(): omp_pause_resource() failed to relinquish
resources on device 0
Warning in .pause_resource(): omp_pause_resource() failed to relinquish
resources on device 0
Warning in .pause_resource(): omp_pause_resource() failed to relinquish
resources on device 0
Warning in .pause_resource(): omp_pause_resource() failed to relinquish
resources on device 0
Warning in .pause_resource(): omp_pause_resource() failed to relinquish
resources on device 0
Warning in .pause_resource(): omp_pause_resource() failed to relinquish
resources on device 0
Warning in .pause_resource(): omp_pause_resource() failed to relinquish
resources on device 0
Warning in .pause_resource(): omp_pause_resource() failed to relinquish
resources on device 0
Warning in .pause_resource(): omp_pause_resource() failed to relinquish
resources on device 0
Warning in .pause_resource(): omp_pause_resource() failed to relinquish
resources on device 0
Warning in .pause_resource(): omp_pause_resource() failed to relinquish
resources on device 0
Warning in .pause_resource(): omp_pause_resource() failed to relinquish
resources on device 0
Warning in .pause_resource(): omp_pause_resource() failed to relinquish
resources on device 0
Warning in .pause_resource(): omp_pause_resource() failed to relinquish
resources on device 0
Warning in .pause_resource(): omp_pause_resource() failed to relinquish
resources on device 0
Warning in .pause_resource(): omp_pause_resource() failed to relinquish
resources on device 0
Warning in .pause_resource(): omp_pause_resource() failed to relinquish
resources on device 0
Warning in .pause_resource(): omp_pause_resource() failed to relinquish
resources on device 0
Warning in .pause_resource(): omp_pause_resource() failed to relinquish
resources on device 0
Warning in .pause_resource(): omp_pause_resource() failed to relinquish
resources on device 0
Warning in .pause_resource(): omp_pause_resource() failed to relinquish
resources on device 0
Warning in .pause_resource(): omp_pause_resource() failed to relinquish
resources on device 0
Warning in .pause_resource(): omp_pause_resource() failed to relinquish
resources on device 0
Warning in .pause_resource(): omp_pause_resource() failed to relinquish
resources on device 0
Warning in .pause_resource(): omp_pause_resource() failed to relinquish
resources on device 0
Warning in .pause_resource(): omp_pause_resource() failed to relinquish
resources on device 0
Warning in .pause_resource(): omp_pause_resource() failed to relinquish
resources on device 0
Warning in .pause_resource(): omp_pause_resource() failed to relinquish
resources on device 0
Warning in .pause_resource(): omp_pause_resource() failed to relinquish
resources on device 0
Warning in .pause_resource(): omp_pause_resource() failed to relinquish
resources on device 0
Warning in .pause_resource(): omp_pause_resource() failed to relinquish
resources on device 0
Warning in .pause_resource(): omp_pause_resource() failed to relinquish
resources on device 0
Warning in .pause_resource(): omp_pause_resource() failed to relinquish
resources on device 0
Warning in .pause_resource(): omp_pause_resource() failed to relinquish
resources on device 0
Warning in .pause_resource(): omp_pause_resource() failed to relinquish
resources on device 0
Warning in .pause_resource(): omp_pause_resource() failed to relinquish
resources on device 0
Warning in .pause_resource(): omp_pause_resource() failed to relinquish
resources on device 0
Warning in .pause_resource(): omp_pause_resource() failed to relinquish
resources on device 0
Warning in .pause_resource(): omp_pause_resource() failed to relinquish
resources on device 0
Warning in .pause_resource(): omp_pause_resource() failed to relinquish
resources on device 0
Warning in .pause_resource(): omp_pause_resource() failed to relinquish
resources on device 0
Warning in .pause_resource(): omp_pause_resource() failed to relinquish
resources on device 0
Warning in .pause_resource(): omp_pause_resource() failed to relinquish
resources on device 0
Warning in .pause_resource(): omp_pause_resource() failed to relinquish
resources on device 0
Warning in .pause_resource(): omp_pause_resource() failed to relinquish
resources on device 0
Warning in .pause_resource(): omp_pause_resource() failed to relinquish
resources on device 0
Warning in .pause_resource(): omp_pause_resource() failed to relinquish
resources on device 0
Warning in .pause_resource(): omp_pause_resource() failed to relinquish
resources on device 0plotReducedDim(sce, "count_PCA", colour_by = "sum")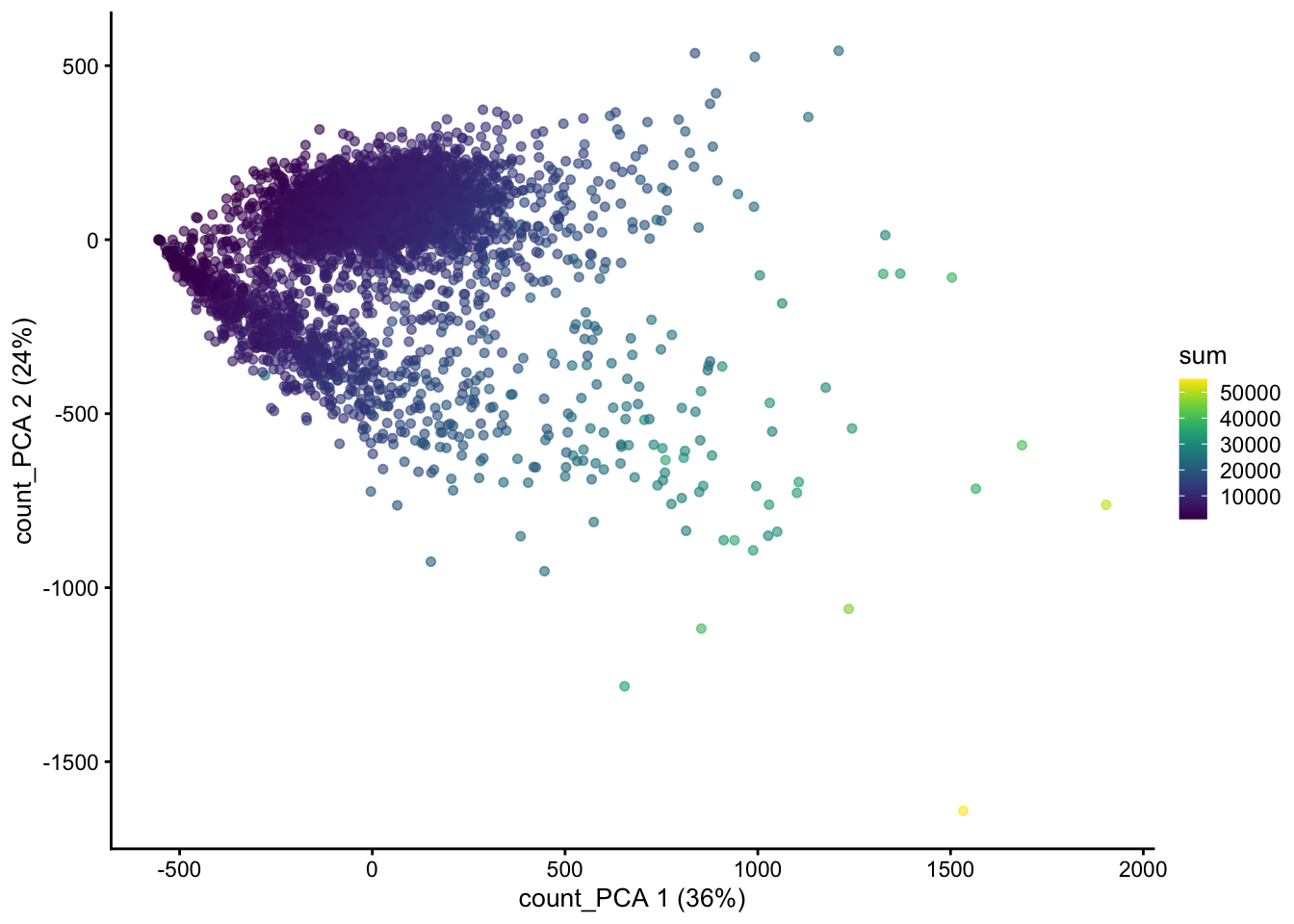
Note that PC1 is correlated with the total UMI counts (sum), meaning that the largest source of variation is related to differences in sequencing depth rather than biological differences between cells.
plot_df <- makePerCellDF(sce, c("count_PCA", "sum"))
ggplot(plot_df, aes(count_PCA.1, sum)) +
geom_point()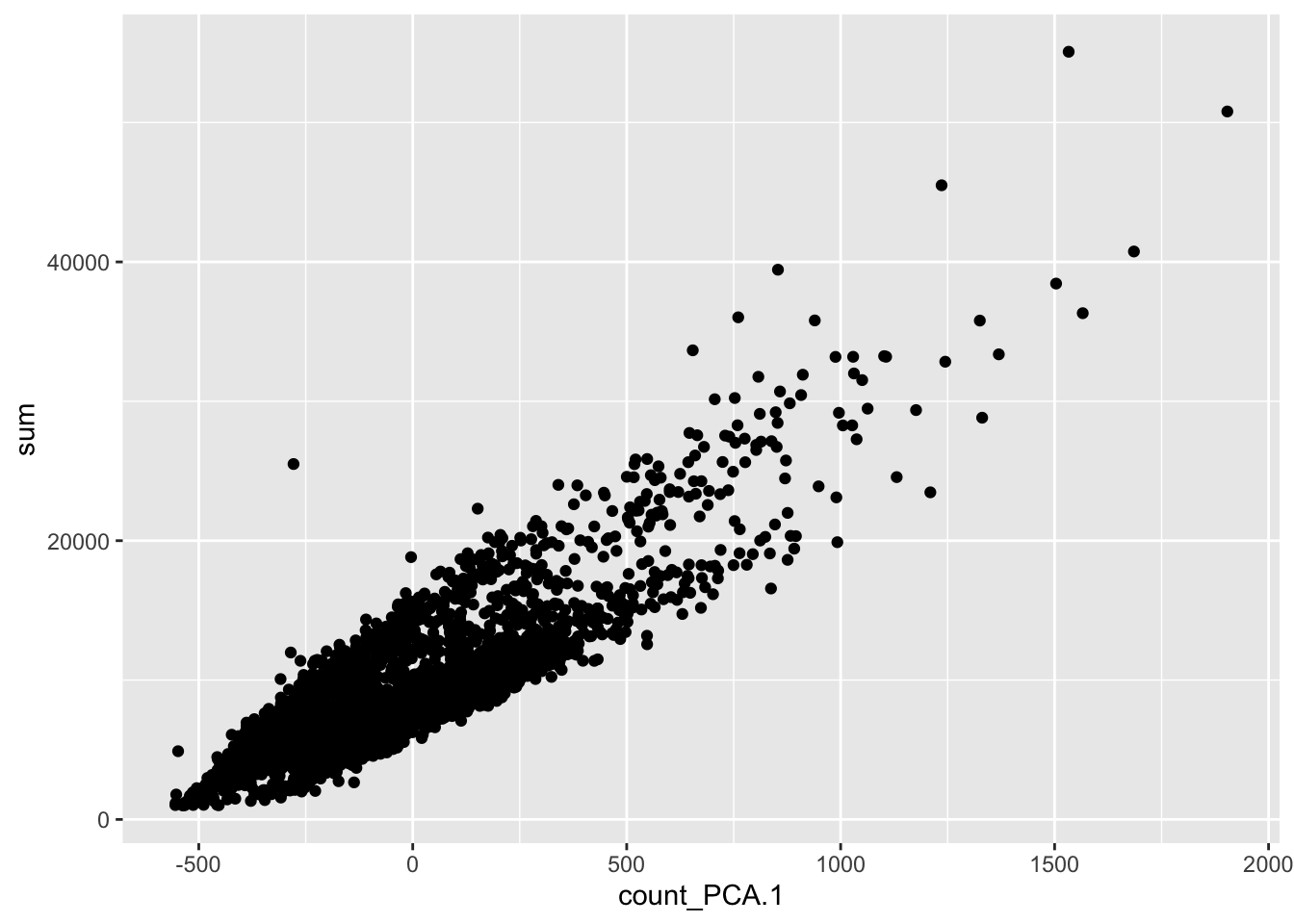
To normalize we will use 3 functions from the scran package. The normalization entails crude clustering to group related cells, identifying a cell-specific normalization factor (size factor), then scaling the counts by this factor and log transforming the data (with a pseudocount). The algorithm is described in detail in the following paper and in the R documentation (?scuttle::computePooledFactors)
Lun AT, Bach K, Marioni JC. Pooling across cells to normalize single-cell RNA sequencing data with many zero counts. Genome Biol. 2016 Apr 27;17:75. doi: 10.1186/s13059-016-0947-7. PMID: 27122128; PMCID: PMC4848819.
set.seed(20231023)
clusters <- quickCluster(sce)
sce <- computeSumFactors(sce, clusters = clusters)
sce <- logNormCounts(sce)set.seed(20231023)
sce <- runPCA(sce, exprs_values = "logcounts", name = "PCA")Warning in .pause_resource(): omp_pause_resource() failed to relinquish
resources on device 0
Warning in .pause_resource(): omp_pause_resource() failed to relinquish
resources on device 0
Warning in .pause_resource(): omp_pause_resource() failed to relinquish
resources on device 0
Warning in .pause_resource(): omp_pause_resource() failed to relinquish
resources on device 0
Warning in .pause_resource(): omp_pause_resource() failed to relinquish
resources on device 0
Warning in .pause_resource(): omp_pause_resource() failed to relinquish
resources on device 0
Warning in .pause_resource(): omp_pause_resource() failed to relinquish
resources on device 0
Warning in .pause_resource(): omp_pause_resource() failed to relinquish
resources on device 0
Warning in .pause_resource(): omp_pause_resource() failed to relinquish
resources on device 0
Warning in .pause_resource(): omp_pause_resource() failed to relinquish
resources on device 0
Warning in .pause_resource(): omp_pause_resource() failed to relinquish
resources on device 0
Warning in .pause_resource(): omp_pause_resource() failed to relinquish
resources on device 0
Warning in .pause_resource(): omp_pause_resource() failed to relinquish
resources on device 0
Warning in .pause_resource(): omp_pause_resource() failed to relinquish
resources on device 0
Warning in .pause_resource(): omp_pause_resource() failed to relinquish
resources on device 0
Warning in .pause_resource(): omp_pause_resource() failed to relinquish
resources on device 0
Warning in .pause_resource(): omp_pause_resource() failed to relinquish
resources on device 0
Warning in .pause_resource(): omp_pause_resource() failed to relinquish
resources on device 0
Warning in .pause_resource(): omp_pause_resource() failed to relinquish
resources on device 0
Warning in .pause_resource(): omp_pause_resource() failed to relinquish
resources on device 0
Warning in .pause_resource(): omp_pause_resource() failed to relinquish
resources on device 0
Warning in .pause_resource(): omp_pause_resource() failed to relinquish
resources on device 0
Warning in .pause_resource(): omp_pause_resource() failed to relinquish
resources on device 0
Warning in .pause_resource(): omp_pause_resource() failed to relinquish
resources on device 0
Warning in .pause_resource(): omp_pause_resource() failed to relinquish
resources on device 0
Warning in .pause_resource(): omp_pause_resource() failed to relinquish
resources on device 0
Warning in .pause_resource(): omp_pause_resource() failed to relinquish
resources on device 0
Warning in .pause_resource(): omp_pause_resource() failed to relinquish
resources on device 0
Warning in .pause_resource(): omp_pause_resource() failed to relinquish
resources on device 0
Warning in .pause_resource(): omp_pause_resource() failed to relinquish
resources on device 0
Warning in .pause_resource(): omp_pause_resource() failed to relinquish
resources on device 0
Warning in .pause_resource(): omp_pause_resource() failed to relinquish
resources on device 0
Warning in .pause_resource(): omp_pause_resource() failed to relinquish
resources on device 0
Warning in .pause_resource(): omp_pause_resource() failed to relinquish
resources on device 0
Warning in .pause_resource(): omp_pause_resource() failed to relinquish
resources on device 0
Warning in .pause_resource(): omp_pause_resource() failed to relinquish
resources on device 0
Warning in .pause_resource(): omp_pause_resource() failed to relinquish
resources on device 0
Warning in .pause_resource(): omp_pause_resource() failed to relinquish
resources on device 0
Warning in .pause_resource(): omp_pause_resource() failed to relinquish
resources on device 0
Warning in .pause_resource(): omp_pause_resource() failed to relinquish
resources on device 0
Warning in .pause_resource(): omp_pause_resource() failed to relinquish
resources on device 0
Warning in .pause_resource(): omp_pause_resource() failed to relinquish
resources on device 0
Warning in .pause_resource(): omp_pause_resource() failed to relinquish
resources on device 0
Warning in .pause_resource(): omp_pause_resource() failed to relinquish
resources on device 0
Warning in .pause_resource(): omp_pause_resource() failed to relinquish
resources on device 0
Warning in .pause_resource(): omp_pause_resource() failed to relinquish
resources on device 0
Warning in .pause_resource(): omp_pause_resource() failed to relinquish
resources on device 0
Warning in .pause_resource(): omp_pause_resource() failed to relinquish
resources on device 0plotReducedDim(sce, "PCA", colour_by = "sum")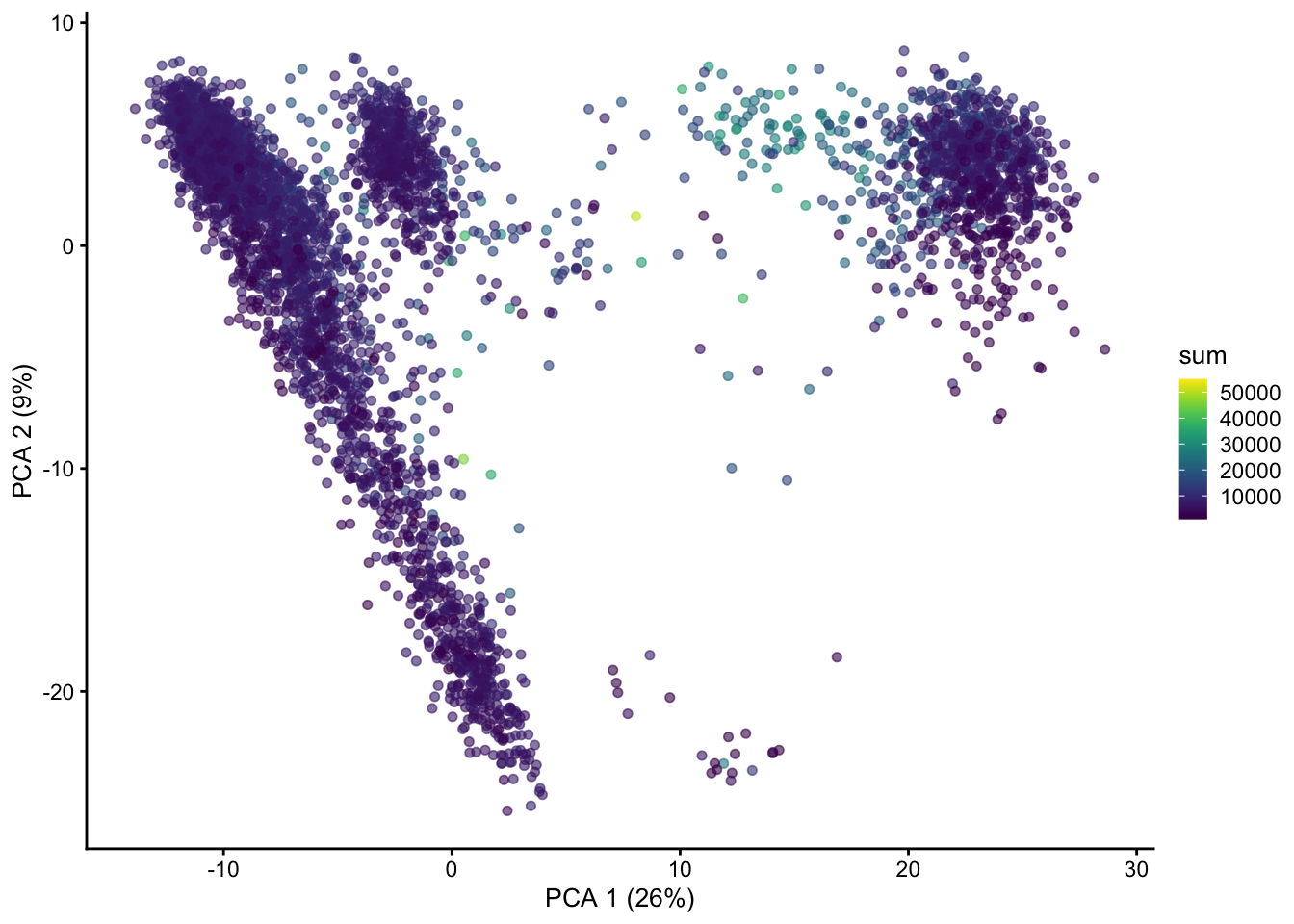
plot_df <- makePerCellDF(sce, c("PCA", "sum"))
ggplot(plot_df, aes(PCA.1, sum)) +
geom_point()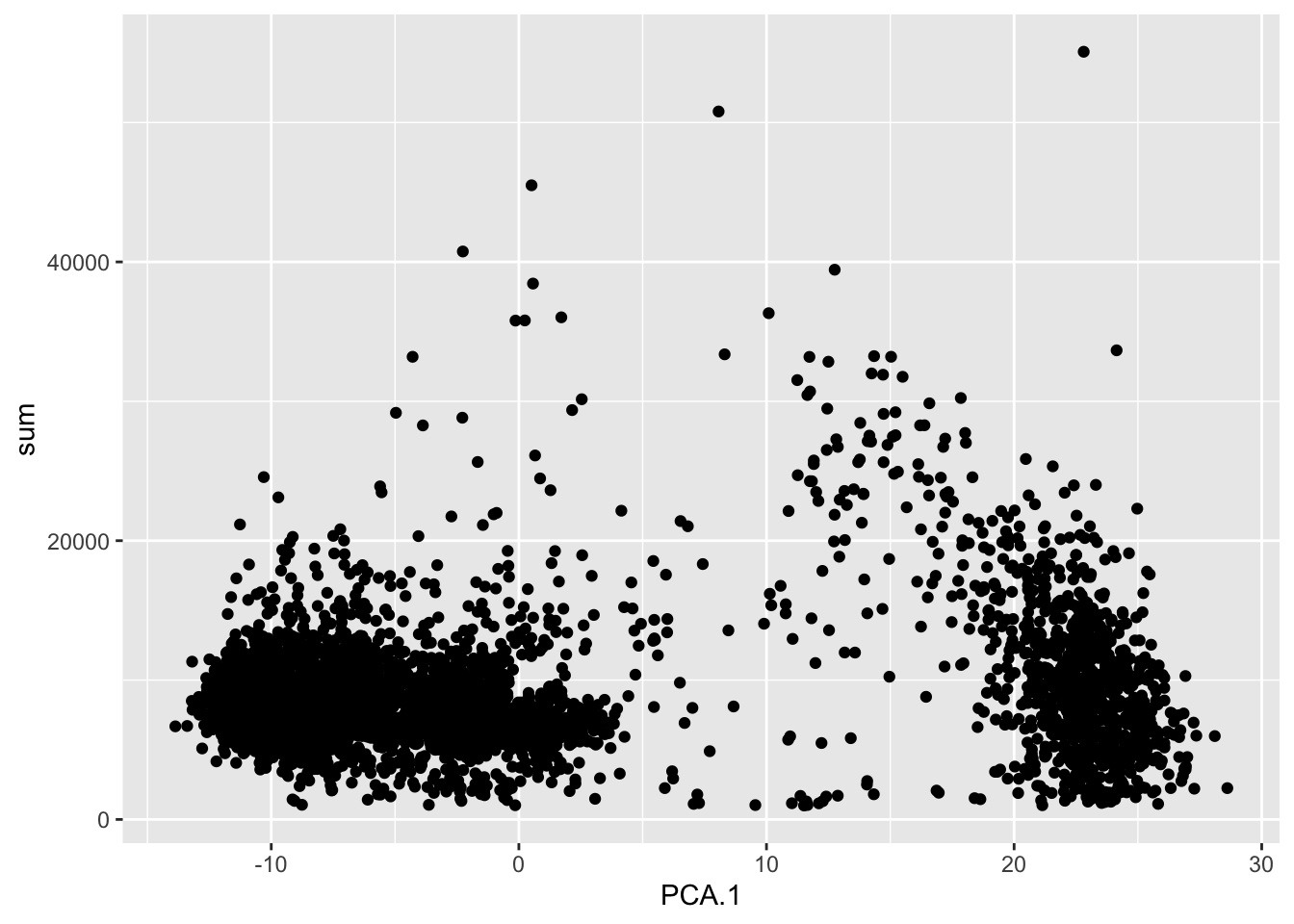
We can now see that PC1 does not correlate with total UMI counts and now shows some more distinct groups of cells, which will likely be different cell populations.