possible_annotations <- builtin_annotations()
# grep to keep those containing "hg19"
hg19_annots <- grep("hg19_genes", possible_annotations, value = T)
# let's keep 5' utr, cds, intron, 3' utr and intergenic
my_hg19_annots <- hg19_annots[c(3,4,7,10,11)]
# build the annotation database
annotations <- build_annotations(genome = 'hg19', annotations = my_hg19_annots)RBP-RNA part 2
mature mRNA regulatory decisions
Just a review of how RBPs control the fate of mRNAs in the cytoplasm.
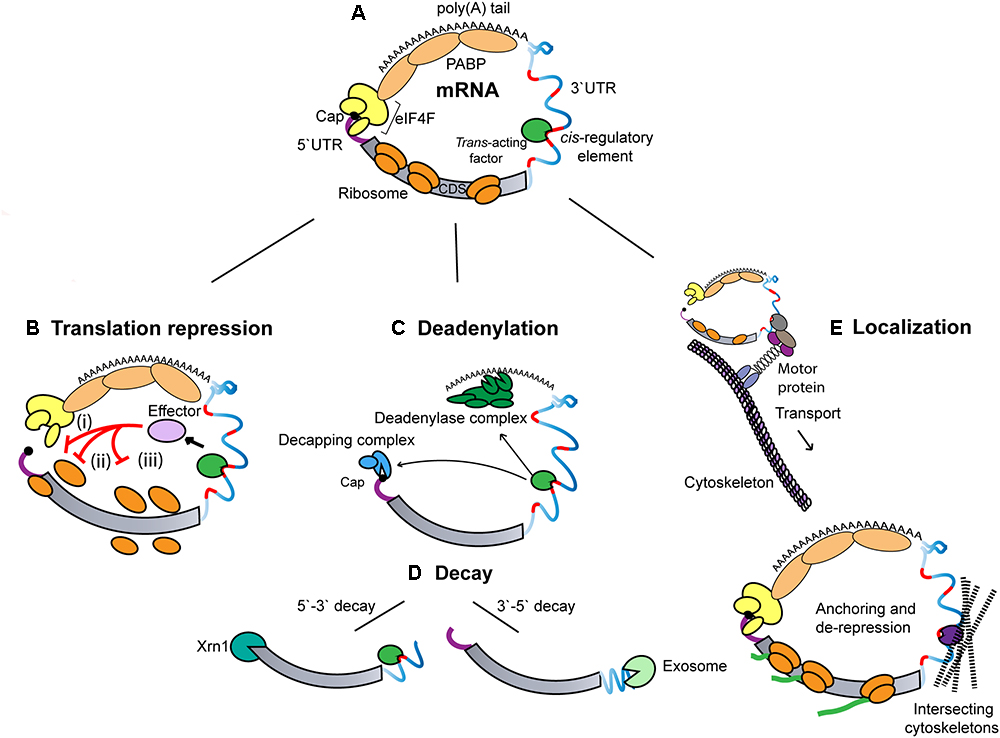
HuR and ARE decay
Remember our analysis of HuR PAR-CLIP data from last class. The model we were testing is that HuR binds to AU-rich elements (ARE) in 3’ UTRs of mRNAs to promote mRNA stability.
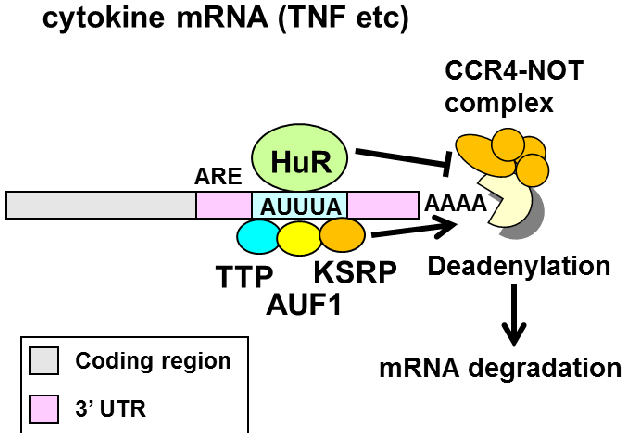 Model predictions:
Model predictions:
- HuR binds to the 3’ UTR and introns.
- HuR binds to AU-rich sequences (AUUUA) and U-rich sequences.
- HuR binding promotes target RNA stabilization (and binding by the other RBPs to the ARE promotes destabilization).
Set up annotation database
Annotate PAR-CLIP data
hur_regions <- read_regions(con = "https://raw.githubusercontent.com/BIMSBbioinfo/RCAS_meta-analysis/master/rbp-binding-sites/SRR248532.clusters.bed",
genome = 'hg19', format = 'bed')
# let's annotate
hur_annot <- annotate_regions(
regions = hur_regions,
annotations = annotations,
ignore.strand = FALSE,
quiet = FALSE) %>%
data.frame()
# keep only columns we need
myInfo <- c("seqnames","start","end","width","strand","annot.symbol","annot.type")
hur_annot <- hur_annot[,myInfo] %>%
unique()
# getting rid of the "hg19_genes_" string to simplify `annot.type`
hur_annot$annot.type <- gsub("hg19_genes_","",hur_annot$annot.type)
table(hur_annot$annot.type)Summarize PAR-CLIP data to gene level
Now we want to get the following info: 1. The # of HuR binding sites per gene. 2. The # of HuR binding sites per region per gene.
# count the # sites per gene and annotation cat
hur_gene_clip <- hur_annot %>%
filter(annot.type!="??") %>%
group_by(??, ??) %>%
dplyr::??() %>%
pivot_wider(names_from = annot.type, values_from = n)
# make NA -> 0
hur_gene_clip <- hur_gene_clip %>% mutate_if(is.numeric , replace_na, replace = 0)
# new column w/total # sites
hur_gene_clip$total <-
# remove symbols that are NA
hur_gene_clip <- hur_gene_clip %>%
filter(annot.symbol != "NA")
# rename cols
colnames(hur_gene_clip)[1] <- "Symbol"
colnames(hur_gene_clip)[3:4] <- c("utr3","utr5")Binding sites per gene
hur_gene_clip_long <- hur_gene_clip %>%
pivot_longer(-??)
colnames(hur_gene_clip_long) <- c("symbol","region","sites")
# histogram of sites/mrna colored by region
ggplot(hur_gene_clip_long %>%
filter(region != "total"),
aes(x=??, fill=??)) +
geom_??() +
scale_x_log10() +
theme_cowplot()Explore intron vs 3’ UTR sites
# how many genes have both intron and/or 3' UTR sites
site_combo <- hur_gene_clip[,2:5] %>%
mutate(
type = case_when(
introns ?? & utr3 ?? ~ "intron_utr3",
introns ?? & utr3 ?? ~ "intron",
introns ?? & utr3 ?? ~ "utr3",
TRUE ~ "other")
)
ggplot(site_combo,
aes(x=??,
fill=??)) +
geom_bar(stat = "count") +
theme_cowplot()HuR binds to both introns and 3’ UTRs.
Genes more often have intronic and 3’ UTR binding sites than either alone.
Load HuR knockdown RNA-seq data
- primary (unspliced precursor) transcripts in
siGFPtreated cells. - mature transcripts in
siGFPtreated cells. - primary (unspliced precursor) transcripts in
siHuRtreated cells. - mature transcripts in
siHuRtreated cells.
# load object called HuR.R
# HuR siRNA RNAseq
??(here("data/block-rna/HuR.R"))
# gene information
gene_info <- read_csv(here("data/block-rna/geneInfo.csv.zip"))
HuR$Gene <- rownames(HuR) # new column gene ids
HuR <- left_join(HuR, gene_info[c(2,11)], by="Gene") # Symbol
# Filter for expression
hur_filt_rnaseq <- HuR %>%
dplyr::filter(rowMeans(HuR[,1:4]) > 1) %>%
dplyr::select(-Gene)integrate siRNA and clip-seq data
#join kd and clip data
kd_clip <- left_join(??,
??,
by = "Symbol")
# convert all NA to 0
kd_clip <- kd_clip %>%
mutate_if(is.numeric,
replace_na,
replace = 0)
# calculate log fold changes
kd_clip <- kd_clip %>%
mutate(
lfc_mature = log2(??) - log2(??),
lfc_primary= log2(??) - log2(??)
)What is the relationship between HuR binding sites and change expression?
Does HuR promote the stability of its mRNA targets?
Does the number of HuR binding influence the degree of stabilization?
Does the region of HuR binding influence stabilization?
To target or not to target?
# create new column for target or not target
kd_clip$target <- case_when(
kd_clip$total > ?? ~ "target",
TRUE ~ "not target")
ggplot(kd_clip,
aes(lfc_mature,
color = ??)) +
stat_??() +
xlim(-2,2) +
theme_cowplot() +
ylab("cumulative fraction of LFC") +
ggtitle("HuR Target vs Not Target") +
geom_hline(yintercept=.75, color="grey") +
geom_hline(yintercept=.5, color="grey") +
geom_hline(yintercept=.25, color="grey")To target a little or a lot?
Does the number of HuR binding influence the degree of stabilization?
# hist(kd_clip$utr3)
# let's make bins for # of sites per 3' UTR
kd_clip$utr3_bin <- cut2(
x = kd_clip$utr3,
c(0,1,2,4,8,1000)
)
# table(kd_clip$utr3_bin)
# let's make bins for UTR
kd_clip$utr3_bin <- recode_factor(kd_clip$utr3_bin,
" 0" = "0",
" 1" = "1",
"[ 2, 4)" = "2-3",
"[ 4, 8)" = "4-7",
"[ 8,1000]" = "8+"
)
ggplot(data = kd_clip,
aes(x = lfc_mature,
color = ??)) +
stat_ecdf() +
xlim(-2,2) +
theme_classic() +
ylab("cumulative fraction- LFCs") +
ggtitle("# of HuR binding sites in 3' UTR") +
geom_hline(yintercept=.75, color="grey", linetype = "dashed") +
geom_hline(yintercept=.5, color="grey", linetype = "dashed") +
geom_hline(yintercept=.25, color="grey", linetype = "dashed") +
geom_segment(aes(x = 0, y = 0, xend = 0, yend = .5, colour = "segment"), color="black", linetype = "dashed")More binding -> more stabilization?
TEST: Does the number of HuR binding influence the degree of stabilization?
# keep only finite values for lfc_mature and lfc_primary
kd_clip_targets <- kd_clip[is.finite(kd_clip$lfc_mature),]
# IMPORTANT relevel indicating what everything will be compared to
kd_clip_targets$utr3_bin <- relevel(factor(kd_clip_targets$utr3_bin), ref = "??")
# calculate fit using `lm`
fit_bins <- lm(data = kd_clip_targets,
formula = ?? ~ ??)
# examine estimates and p-vals
tidy(fit_bins) %>%
gt()More binding -> more stabilization?
TEST: Does the number of HuR binding influence the degree of stabilization?
# mean lfc of mRNAs with no 3' UTR binding sites
ref_mean <- kd_clip_targets %>%
filter(utr3_bin == "0") %>%
pull(lfc_mature) %>%
mean()
# examine estimates and color by p-vals
tidy(fit_bins) %>%
ggplot(
aes(x=??,
y=??,
fill=??
)
) +
geom_bar(stat="identity") +
coord_flip() +
geom_hline(yintercept = ref_mean,
color = "red") +
theme_cowplot()