Plot repertoire diversity
Usage
plot_diversity(
input,
data_col,
cluster_col = NULL,
group_col = NULL,
method = abdiv::simpson,
downsample = FALSE,
n_boots = 0,
chain = NULL,
chain_col = global$chain_col,
sep = global$sep,
plot_colors = NULL,
plot_lvls = names(plot_colors),
panel_nrow = NULL,
panel_scales = "free",
n_label = NULL,
p_label = "all",
p_method = NULL,
p_file = NULL,
label_params = list(),
...
)Arguments
- input
Single cell object or data.frame containing V(D)J data. If a data.frame is provided, the cell barcodes should be stored as row names.
- data_col
meta.data column containing values to use for calculating diversity, e.g. 'clonotype_id'
- cluster_col
meta.data column containing cluster IDs to use for grouping cells when calculating clonotype abundance
- group_col
meta.data column to use for grouping clusters present in cluster_col
- method
Function to use for calculating diversity, e.g. abdiv::simpson. A named list of functions can be passed to plot multiple diversity metrics, e.g. list(simpson = abdiv::simpson, shannon = abdiv::shannon)
- downsample
Downsample clusters to the same size when calculating diversity metrics
- n_boots
Number of bootstrap replicates for calculating standard deviation, if n_boots is 0 this will be skipped.
- chain
Chain to use for calculating diversity. To calculate diversity for a single chain, the column passed to the data_col argument must contain per-chain data such as CDR3 sequences. Set to NULL to include all chains.
- chain_col
meta.data column containing chains for each cell
- sep
Separator used for storing per-chain V(D)J data for each cell
- plot_colors
Character vector containing colors for plotting
- plot_lvls
Character vector containing levels for ordering
- panel_nrow
The number of rows to use for arranging plot panels
- panel_scales
Should scales for plot panels be fixed or free. This passes a scales specification to ggplot2::facet_wrap, can be 'fixed', 'free', 'free_x', or 'free_y'. 'fixed' will cause panels to share the same scales.
- n_label
Location on plot where n label should be added, this can be any combination of the following:
'corner', display the total number of cells plotted in the top right corner, the position of the label can be modified by passing
xandyspecifications with thelabel_paramsargument'axis', display the number of cells plotted for each group shown on the x-axis
'legend', display the number of cells plotted for each group shown in the plot legend
'none', do not display the number of cells plotted
- p_label
Specification indicating how p-values should be labeled on plot, this can one of the following:
'none', do not display p-values
'all', show p-values for all groups
A named vector providing p-value cutoffs and labels to display, e.g.
c('*' = 0.05, '**' = 0.01, '***' = 0.001). The keyword 'value' can be used to display the p-value for those less than a certain cutoff, e.g.c(value = 0.05, ns = 1.1)will show significant p-values, all others will be labeled 'ns'.
- p_method
Method to use for calculating p-values, by default when comparing two groups a t-test will be used. When comparing more than two groups the Kruskal-Wallis test will be used. p-values are adjusted for multiple testing using Bonferroni correction. Possible methods include:
't', two sample t-test performed with
stats::t.test()'wilcox', Wilcoxon rank sum test performed with
stats::wilcox.test()'kruskal', Kruskal-Wallis test performed with
stats::kruskal.test()
- p_file
File path to save table containing p-values for each comparison.
- label_params
Named list providing additional parameters to modify n label aesthetics, e.g. list(size = 4, color = "red")
- ...
Additional arguments to pass to ggplot2, e.g. color, fill, size, linetype, etc.
Examples
# Specify method to use for calculating repertoire diversity
plot_diversity(
vdj_sce,
data_col = "clonotype_id",
method = abdiv::shannon
)
 # Plot diversity separately for each cell cluster
plot_diversity(
vdj_sce,
data_col = "clonotype_id",
cluster_col = "orig.ident"
)
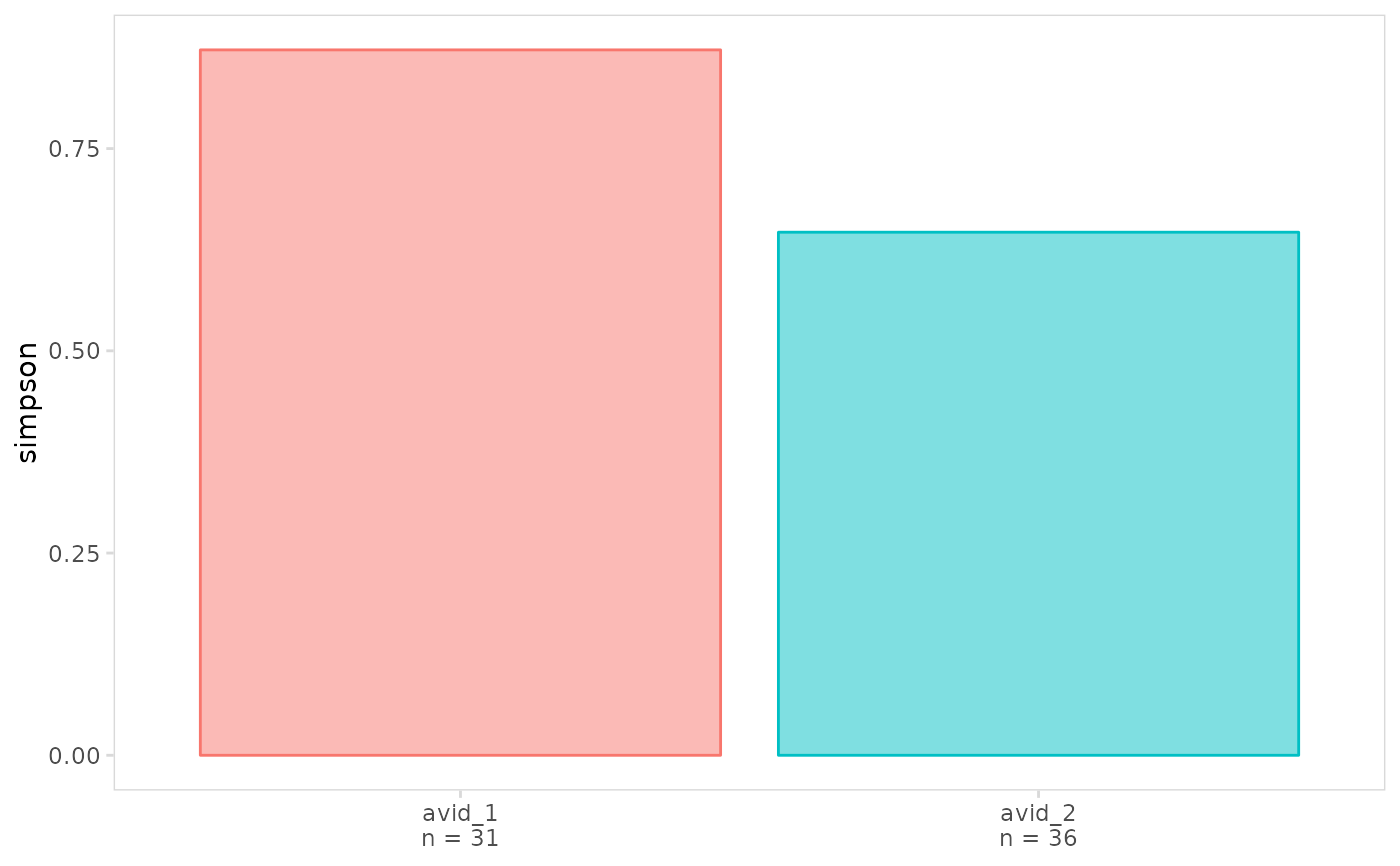
# Plot diversity separately for each cell cluster
plot_diversity(
vdj_sce,
data_col = "clonotype_id",
cluster_col = "orig.ident"
)
 # Plot multiple diversity metrics
mets <- list(
simpson = abdiv::simpson,
shannon = abdiv::shannon
)
plot_diversity(
vdj_sce,
data_col = "clonotype_id",
cluster_col = "orig.ident",
method = mets
)
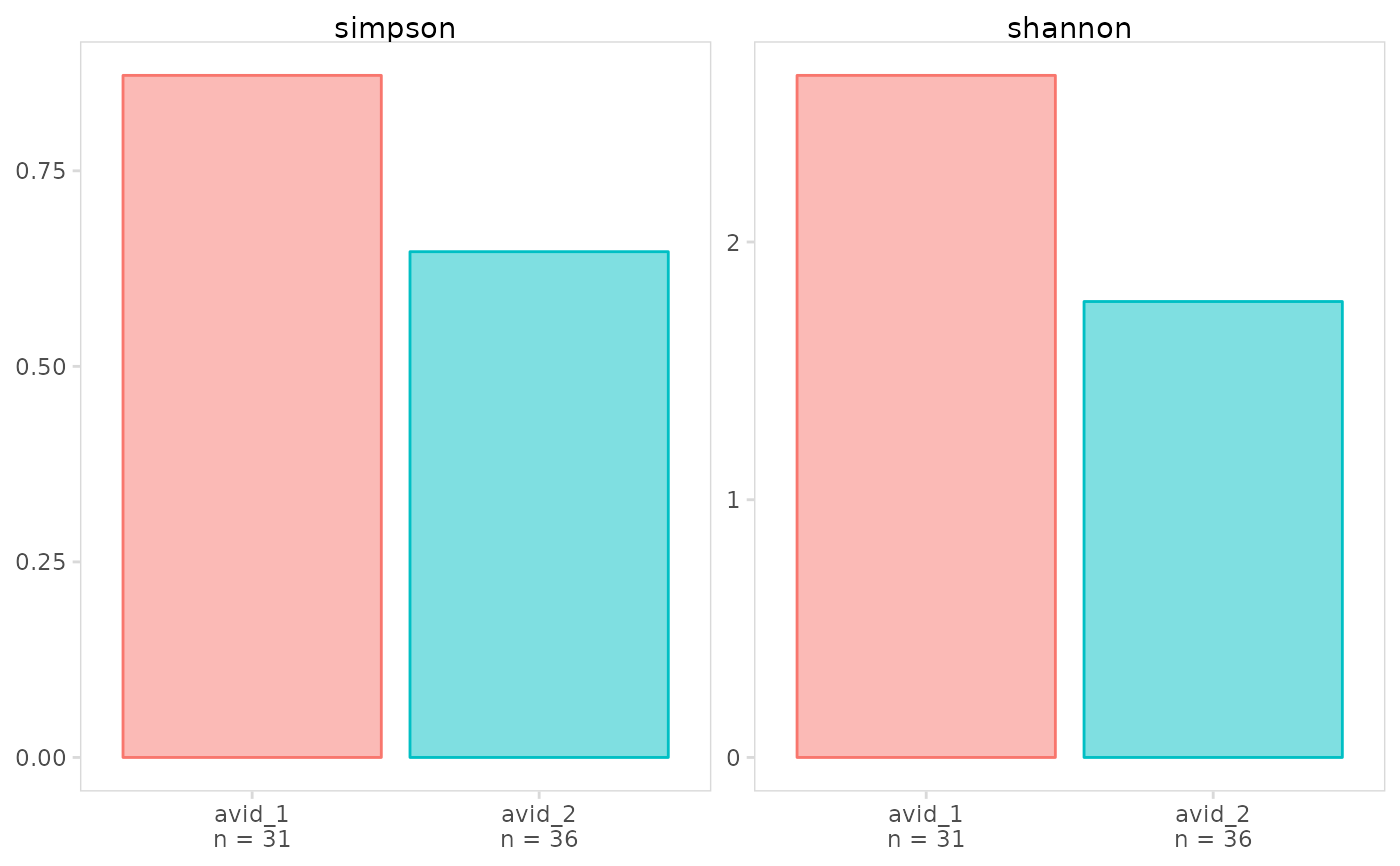
# Plot multiple diversity metrics
mets <- list(
simpson = abdiv::simpson,
shannon = abdiv::shannon
)
plot_diversity(
vdj_sce,
data_col = "clonotype_id",
cluster_col = "orig.ident",
method = mets
)
 # Specify how to organize panels when plotting multiple metrics
plot_diversity(
vdj_sce,
data_col = "clonotype_id",
method = mets,
panel_nrow = 2
)
# Specify how to organize panels when plotting multiple metrics
plot_diversity(
vdj_sce,
data_col = "clonotype_id",
method = mets,
panel_nrow = 2
)
