Increase the size of input intervals.
Usage
bed_slop(
x,
genome,
both = 0,
left = 0,
right = 0,
fraction = FALSE,
strand = FALSE,
trim = FALSE,
...
)Arguments
- x
- genome
- both
number of bases on both sizes
- left
number of bases on left side
- right
number of bases on right side
- fraction
define flanks based on fraction of interval length
- strand
define
leftandrightbased on strand- trim
adjust coordinates for out-of-bounds intervals
- ...
extra arguments (not used)
See also
https://bedtools.readthedocs.io/en/latest/content/tools/slop.html
Other single set operations:
bed_cluster(),
bed_complement(),
bed_flank(),
bed_genomecov(),
bed_merge(),
bed_partition(),
bed_shift()
Examples
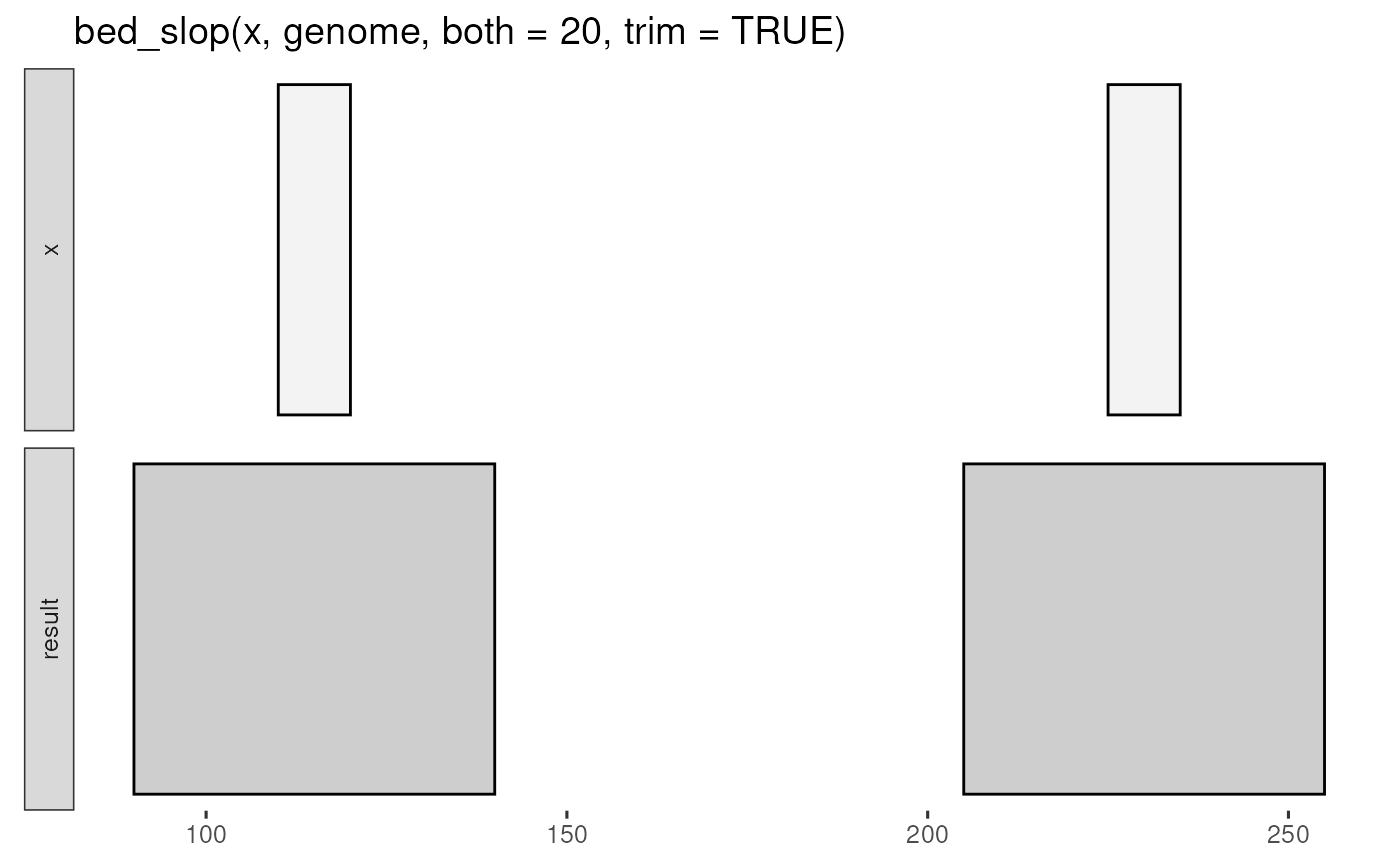
x <- tibble::tribble(
~chrom, ~start, ~end,
"chr1", 110, 120,
"chr1", 225, 235
)
genome <- tibble::tribble(
~chrom, ~size,
"chr1", 400
)
bed_glyph(bed_slop(x, genome, both = 20, trim = TRUE))
 genome <- tibble::tribble(
~chrom, ~size,
"chr1", 5000
)
x <- tibble::tribble(
~chrom, ~start, ~end, ~name, ~score, ~strand,
"chr1", 500, 1000, ".", ".", "+",
"chr1", 1000, 1500, ".", ".", "-"
)
bed_slop(x, genome, left = 100)
#> # A tibble: 2 × 6
#> chrom start end name score strand
#> <chr> <dbl> <dbl> <chr> <chr> <chr>
#> 1 chr1 400 1000 . . +
#> 2 chr1 900 1500 . . -
bed_slop(x, genome, right = 100)
#> # A tibble: 2 × 6
#> chrom start end name score strand
#> <chr> <dbl> <dbl> <chr> <chr> <chr>
#> 1 chr1 500 1100 . . +
#> 2 chr1 1000 1600 . . -
bed_slop(x, genome, both = 100)
#> # A tibble: 2 × 6
#> chrom start end name score strand
#> <chr> <dbl> <dbl> <chr> <chr> <chr>
#> 1 chr1 400 1100 . . +
#> 2 chr1 900 1600 . . -
bed_slop(x, genome, both = 0.5, fraction = TRUE)
#> # A tibble: 2 × 6
#> chrom start end name score strand
#> <chr> <dbl> <dbl> <chr> <chr> <chr>
#> 1 chr1 250 1250 . . +
#> 2 chr1 750 1750 . . -
genome <- tibble::tribble(
~chrom, ~size,
"chr1", 5000
)
x <- tibble::tribble(
~chrom, ~start, ~end, ~name, ~score, ~strand,
"chr1", 500, 1000, ".", ".", "+",
"chr1", 1000, 1500, ".", ".", "-"
)
bed_slop(x, genome, left = 100)
#> # A tibble: 2 × 6
#> chrom start end name score strand
#> <chr> <dbl> <dbl> <chr> <chr> <chr>
#> 1 chr1 400 1000 . . +
#> 2 chr1 900 1500 . . -
bed_slop(x, genome, right = 100)
#> # A tibble: 2 × 6
#> chrom start end name score strand
#> <chr> <dbl> <dbl> <chr> <chr> <chr>
#> 1 chr1 500 1100 . . +
#> 2 chr1 1000 1600 . . -
bed_slop(x, genome, both = 100)
#> # A tibble: 2 × 6
#> chrom start end name score strand
#> <chr> <dbl> <dbl> <chr> <chr> <chr>
#> 1 chr1 400 1100 . . +
#> 2 chr1 900 1600 . . -
bed_slop(x, genome, both = 0.5, fraction = TRUE)
#> # A tibble: 2 × 6
#> chrom start end name score strand
#> <chr> <dbl> <dbl> <chr> <chr> <chr>
#> 1 chr1 250 1250 . . +
#> 2 chr1 750 1750 . . -